

Description
Craniometaphyseal dysplasia is a rare condition characterized by thickening (overgrowth) of bones in the skull (cranium) and abnormalities in a region at the end of long bones known as the metaphysis. The abnormal bone growth continues throughout life. Except in the most severe cases, the lifespan of people with craniometaphyseal dysplasia is normal.
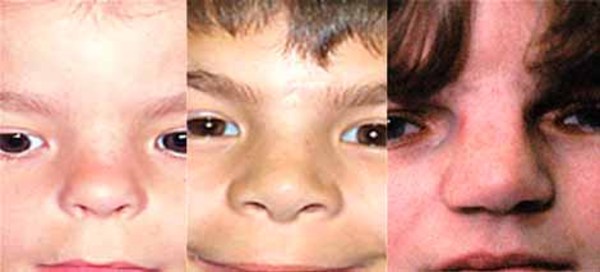
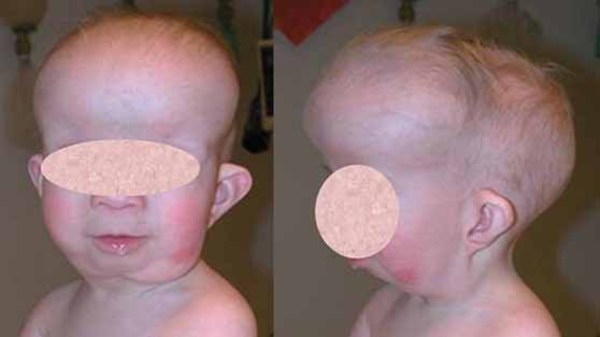
Bone overgrowth in the head causes many of the signs and symptoms of craniometaphyseal dysplasia. Affected individuals typically have distinctive facial features such as a wide nasal bridge, a prominent forehead, wide-set eyes (hypertelorism), and a prominent jaw. Excess bone formation in the jaw can delay teething (dentition) or result in absent (non-erupting) teeth. Infants with craniometaphyseal dysplasia may have breathing or feeding problems caused by narrow nasal passages. In severe cases, abnormal bone growth can pinch (compress) the nerves that extend from the brain to various areas of the head and neck (cranial nerves). Compression of the cranial nerves can lead to paralyzed facial muscles (facial nerve palsy), blindness, or deafness.

The x-rays of individuals with craniometaphyseal dysplasia show unusually shaped long bones, particularly long bones in the legs. The ends of these bones are wider and appear less dense than usual in people with this condition.
There are two types of craniometaphyseal dysplasia, which are distinguished by their pattern of inheritance and genetic cause. They are known as the autosomal dominant and autosomal recessive types.
Frequency
Craniometaphyseal dysplasia is a very rare disorder; its incidence is unknown.
Causes
Mutations in the ANKH gene cause autosomal dominant craniometaphyseal dysplasia. The ANKH gene provides instructions for making a protein that plays a role in the development and function of cells that build bones (osteoblasts) and cells that break down bone (osteoclasts). Osteoclasts are involved in bone remodeling, a normal process in which old bone is removed and new bone is created to replace it. In addition, the ANKH protein transports a molecule called pyrophosphate out of cells. The pyrophosphate found outside of cells (extracellular pyrophosphate) helps control bone formation by preventing mineralization, the process by which minerals such as calcium and phosphorus are deposited in developing bones. The ANKH protein may have other, unknown functions.
Mutations in the ANKH gene that cause autosomal dominant craniometaphyseal dysplasia impair the maturation (differentiation) of osteoclasts, which likely disrupts bone remodeling. Reduced breakdown of bone tissue can contribute to the bone thickening characteristic of craniometaphyseal dysplasia. ANKH gene mutations may also reduce the protein's ability to transport pyrophosphate out of cells. A shortage of extracellular pyrophosphate can increase bone mineralization, which may also contribute to the bone abnormalities.
A mutation in the GJA1 gene causes some cases of autosomal recessive craniometaphyseal dysplasia. This gene provides instructions for making a protein called connexin 43, which is involved in the development of many tissues in the body, including bone. The protein may be involved in bone remodeling. It is unclear how a mutation in the GJA1 gene leads to the particular bone abnormalities of craniometaphyseal dysplasia.
The genetic cause of many cases of autosomal recessive craniometaphyseal dysplasia is unknown. It is likely that other, unidentified genes are involved in this form of the disorder.
Inheritance
When caused by mutations in the ANKH gene, craniometaphyseal dysplasia follows an autosomal dominant pattern, which means one altered copy of the ANKH gene in each cell is sufficient to cause the disorder. Individuals with autosomal dominant craniometaphyseal dysplasia typically have one parent who also has the condition. Less often, cases result from new mutations in the gene and occur in people with no history of the disorder in their family.

When caused by mutations in the GJA1 gene, craniometaphyseal dysplasia has an autosomal recessive inheritance pattern, which means both copies of the GJA1 gene in each cell are altered. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the disorder.

Other Names for This Condition
- Autosomal dominant craniometaphyseal dysplasia
- Autosomal recessive craniometaphyseal dysplasia
- CMD
- CMDD
- CMDJ
- CMDR
- Craniometaphyseal dysplasia, Jackson type
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Addison WN, Azari F, Sorensen ES, Kaartinen MT, McKee MD. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem. 2007 May 25;282(21):15872-83. doi: 10.1074/jbc.M701116200. Epub 2007 Mar 23. Citation on PubMed
- Hu Y, Chen IP, de Almeida S, Tiziani V, Do Amaral CM, Gowrishankar K, Passos-Bueno MR, Reichenberger EJ. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS One. 2013 Aug 12;8(8):e73576. doi: 10.1371/journal.pone.0073576. eCollection 2013. Citation on PubMed or Free article on PubMed Central
- Iughetti P, Alonso LG, Wilcox W, Alonso N, Passos-Bueno MR. Mapping of the autosomal recessive (AR) craniometaphyseal dysplasia locus to chromosome region 6q21-22 and confirmation of genetic heterogeneity for mild AR spondylocostal dysplasia. Am J Med Genet. 2000 Dec 18;95(5):482-91. doi: 10.1002/1096-8628(20001218)95:53.0.co;2-x. Citation on PubMed
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001 May;28(1):37-41. doi: 10.1038/ng0501-37. Citation on PubMed
- Reichenberger E, Chen IP. Autosomal Dominant Craniometaphyseal Dysplasia. 2007 Aug 27 [updated 2025 Aug 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1461/ Citation on PubMed
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, Gardner J, Hamersma H, Sellars S, Ramesar R, Lidral AC, Sommer A, Raposo do Amaral CM, Gorlin RJ, Mulliken JB, Olsen BR. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001 Jun;68(6):1321-6. doi: 10.1086/320612. Epub 2001 Apr 16. Citation on PubMed or Free article on PubMed Central
- Talbot J, Brion R, Lamora A, Mullard M, Morice S, Heymann D, Verrecchia F. Connexin43 intercellular communication drives the early differentiation of human bone marrow stromal cells into osteoblasts. J Cell Physiol. 2018 Feb;233(2):946-957. doi: 10.1002/jcp.25938. Epub 2017 May 23. Citation on PubMed
- Zaka R, Williams CJ. Role of the progressive ankylosis gene in cartilage mineralization. Curr Opin Rheumatol. 2006 Mar;18(2):181-6. doi: 10.1097/01.bor.0000209432.36355.6e. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.