

Description
Craniofrontonasal syndrome is a rare condition characterized by the premature closure of certain bones of the skull (craniosynostosis) during development, which affects the shape of the head and face. The condition is named for the areas of the body that are typically affected: the skull (cranio-), face (fronto-), and nose (nasal).
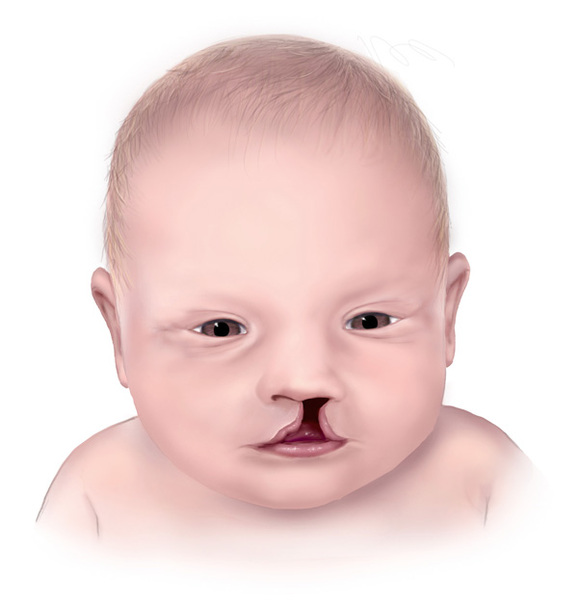
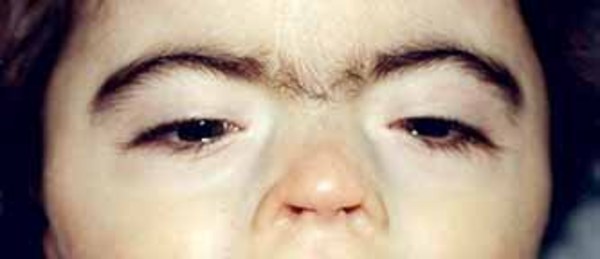
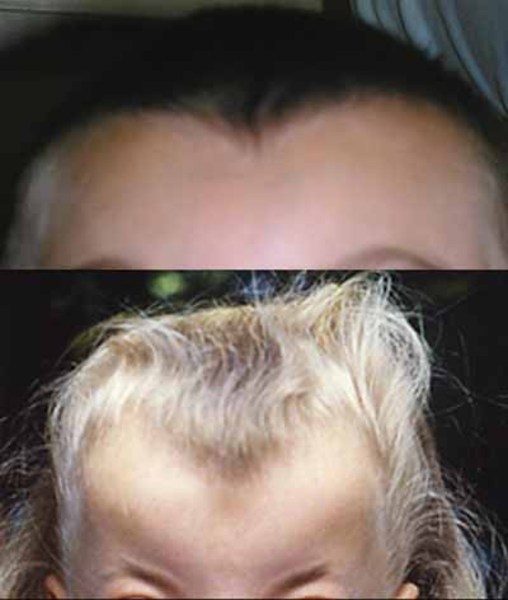
In people with craniofrontonasal syndrome, the skull bones along the coronal suture, which is the growth line that goes over the head from ear to ear, closes early. These changes can result in an abnormally shaped head and distinctive facial features. The size and shape of facial structures may differ between the right and left sides of the face (facial asymmetry) in individuals with craniofrontonasal syndrome. Affected individuals may also have wide-set eyes (ocular hypertelorism), eyes that do not point in the same direction (strabismus), involuntary eye movements (nystagmus), a slit (cleft) in the tip of the nose, a wide nasal bridge, an upper lip that points outward (called a tented lip), or a cleft in the upper lip with or without a cleft in roof of the mouth (palate). Some affected individuals have brain abnormalities, such as absent or underdeveloped tissue connecting the left and right halves of the brain (agenesis or dysgenesis of the corpus callosum). However, intelligence is usually unaffected in people with this condition. Females with craniofrontonasal syndrome typically have more severe signs and symptoms than affected males, who often have hypertelorism and rarely, cleft lip.


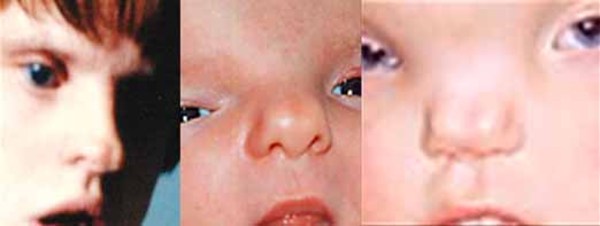
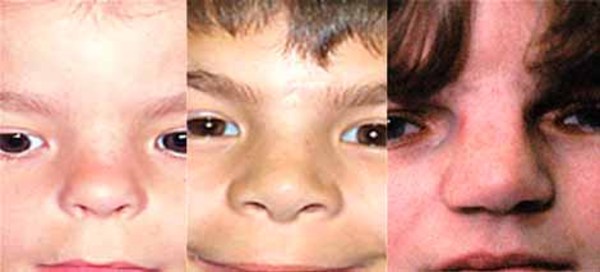


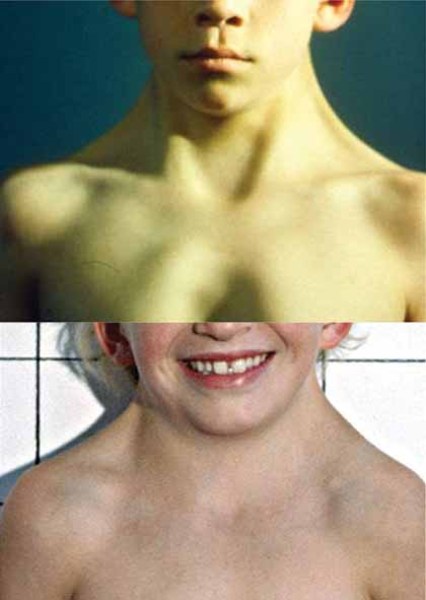
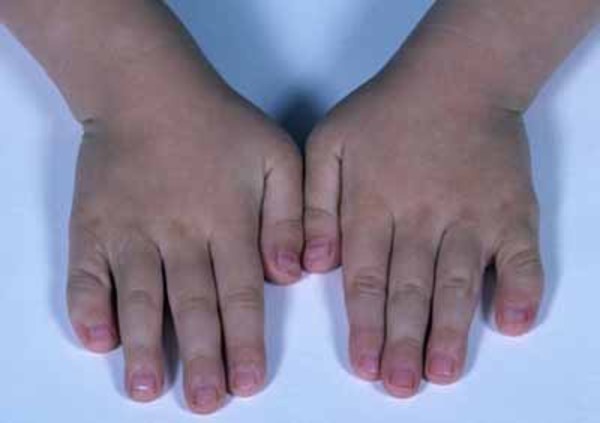
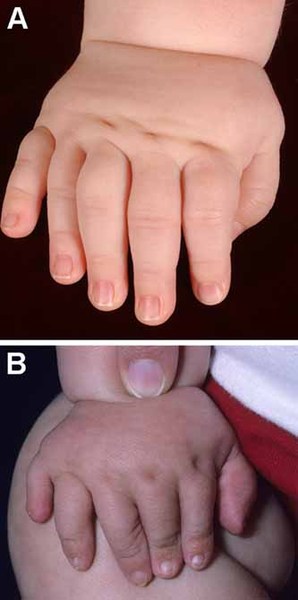
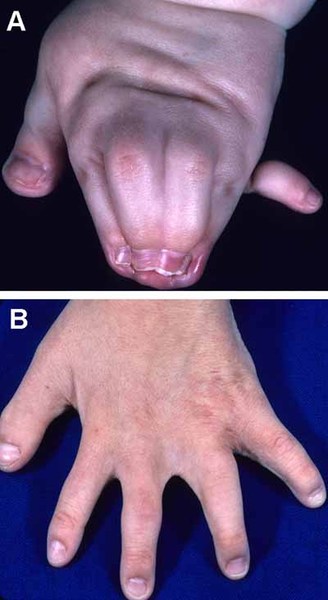
Other common features of craniofrontonasal syndrome include extra folds of skin on the neck (webbed neck), ridged nails, unusual curving of the fingers or toes (clinodactyly), extra fingers (polydactyly) or fingers that are fused together (syndactyly), low-set breasts, a sunken chest (pectus excavatum), a spine that curves to the side (scoliosis), or narrow and sloped shoulders with reduced range of motion. People with this condition may also have eyebrows that grow together in the middle (synophrys), a widow's peak hairline with a low hairline in the back, or wiry hair.

Frequency
Craniofrontonasal syndrome is a very rare condition, although its prevalence is unknown. More than 115 cases have been described in the scientific literature.
Causes
Craniofrontonasal syndrome is caused by mutations in a gene known as EFNB1. This gene provides instructions for making a protein called ephrin B1. This protein spans the cell membrane where it can attach (bind) to other proteins on the surface of neighboring cells. Protein binding between nearby cells helps cells stick to one another (cell adhesion) and communicate, which are important for the normal shaping (patterning) of many tissues and organs before birth.
Mutations in the EFNB1 gene result in a shortage (deficiency) of ephrin B1 protein. Most of these mutations lead to an abnormally short version of the molecule that acts as the genetic blueprint for the ephrin B1 protein. The shortened molecules are quickly broken down before protein can be made. A deficiency of ephrin B1 protein prevents cell adhesion, which disrupts normal patterning in tissues before birth, leading to the signs and symptoms of craniofrontonasal syndrome.
Inheritance
Craniofrontonasal syndrome is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. Males have only one X chromosome and females have two copies of the X chromosome. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

Researchers suspect that the signs and symptoms of craniofrontonasal syndrome vary in severity between males and females in part because of a normal process called X-inactivation. Early in embryonic development in females, one of the two X chromosomes is permanently turned off (inactivated) in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, so that each X chromosome is active in about half the body's cells. This means that in affected females with craniofrontonasal syndrome, the X chromosome with an EFNB1 gene mutation is active in about half of cells, and the X chromosome with the normal EFNB1 gene is active in about half. Because X-inactivation leads to some cells that produce functional ephrin B1 protein and some cells that do not, patterning of tissues becomes patchy, which can alter development of the head and face.
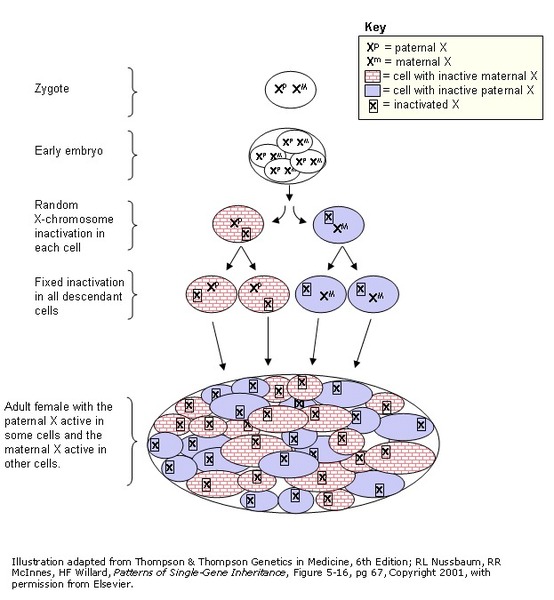
In affected males, all cells have a single X chromosome with an EFNB1 gene mutation. Because ephrin B1 activity is completely missing in all cells, normal tissue patterning cannot occur and it is thought that other proteins perform similar functions to compensate.
Other Names for This Condition
- CFND
- CFNS
- Craniofrontonasal dysplasia
- Craniofrontonasal dystosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Howaldt A, Nampoothiri S, Yesodharan D, Udayakumaran S, Subash P, Kornak U. Four novel mutations in EFNB1 in Indian patients with craniofrontonasal syndrome. J Hum Genet. 2019 Sep;64(9):867-873. doi: 10.1038/s10038-019-0638-9. Epub 2019 Jul 8. Citation on PubMed
- Niethamer TK, Larson AR, O'Neill AK, Bershteyn M, Hsiao EC, Klein OD, Pomerantz JH, Bush JO. EPHRIN-B1 Mosaicism Drives Cell Segregation in Craniofrontonasal Syndrome hiPSC-Derived Neuroepithelial Cells. Stem Cell Reports. 2017 Mar 14;8(3):529-537. doi: 10.1016/j.stemcr.2017.01.017. Epub 2017 Feb 23. Citation on PubMed or Free article on PubMed Central
- Romanelli Tavares VL, Kague E, Musso CM, Alegria TGP, Freitas RS, Bertola DR, Twigg SRF, Passos-Bueno MR. Craniofrontonasal Syndrome Caused by Introduction of a Novel uATG in the 5'UTR of EFNB1. Mol Syndromol. 2019 Feb;10(1-2):40-47. doi: 10.1159/000490635. Epub 2018 Jul 3. Citation on PubMed or Free article on PubMed Central
- Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, Morriss-Kay GM, Wilkie AO. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. 2004 Jun 8;101(23):8652-7. doi: 10.1073/pnas.0402819101. Epub 2004 May 27. Citation on PubMed or Free article on PubMed Central
- van den Elzen ME, Twigg SR, Goos JA, Hoogeboom AJ, van den Ouweland AM, Wilkie AO, Mathijssen IM. Phenotypes of craniofrontonasal syndrome in patients with a pathogenic mutation in EFNB1. Eur J Hum Genet. 2014 Aug;22(8):995-1001. doi: 10.1038/ejhg.2013.273. Epub 2013 Nov 27. Citation on PubMed or Free article on PubMed Central
- Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, Adams RH, Wieacker P. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet. 2004 Jun;74(6):1209-15. doi: 10.1086/421532. Epub 2004 Apr 29. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.