

Description
Craniofacial microsomia is a term used to describe a spectrum of abnormalities that primarily affect the development of the skull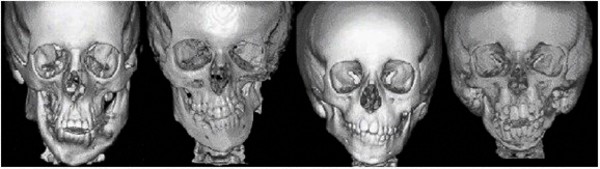 (cranium) and face before birth. Microsomia means abnormal smallness of body structures. Most people with craniofacial microsomia have differences in the size and shape of facial structures between the right and left sides of the face (facial asymmetry). In about two-thirds of cases, both sides of the face have abnormalities, which usually differ from one side to the other. Other individuals with craniofacial microsomia are affected on only one side of the face. The facial characteristics in craniofacial microsomia typically include underdevelopment of one side of the upper or lower jaw (maxillary or mandibular hypoplasia), which can cause dental problems and difficulties with feeding and speech. In cases of severe mandibular hypoplasia, breathing may also be affected.
(cranium) and face before birth. Microsomia means abnormal smallness of body structures. Most people with craniofacial microsomia have differences in the size and shape of facial structures between the right and left sides of the face (facial asymmetry). In about two-thirds of cases, both sides of the face have abnormalities, which usually differ from one side to the other. Other individuals with craniofacial microsomia are affected on only one side of the face. The facial characteristics in craniofacial microsomia typically include underdevelopment of one side of the upper or lower jaw (maxillary or mandibular hypoplasia), which can cause dental problems and difficulties with feeding and speech. In cases of severe mandibular hypoplasia, breathing may also be affected.
People with craniofacial microsomia usually have ear abnormalities affecting one or both ears, typically to different degrees . They may have growths of skin (skin tags) in front of the ear (preauricular tags), an underdeveloped or absent external ear (microtia or anotia), or a closed or absent ear canal; these abnormalities may lead to hearing loss. Eye problems are less common in craniofacial microsomia, but some affected individuals have an unusually small eyeball (microphthalmia) or other eye abnormalities that result in vision loss.
. They may have growths of skin (skin tags) in front of the ear (preauricular tags), an underdeveloped or absent external ear (microtia or anotia), or a closed or absent ear canal; these abnormalities may lead to hearing loss. Eye problems are less common in craniofacial microsomia, but some affected individuals have an unusually small eyeball (microphthalmia) or other eye abnormalities that result in vision loss.
Abnormalities in other parts of the body, such as malformed bones of the spine (vertebrae), abnormally shaped kidneys, and heart defects, may also occur in people with craniofacial microsomia.
Many other terms have been used for craniofacial microsomia. These other names generally refer to forms of craniofacial microsomia with specific combinations of signs and symptoms, although sometimes they are used interchangeably. Hemifacial microsomia often refers to craniofacial microsomia with maxillary or mandibular hypoplasia. People with hemifacial microsomia and noncancerous (benign) growths in the eye called epibulbar dermoids may be said to have Goldenhar syndrome or oculoauricular dysplasia.
Frequency
Craniofacial microsomia has been estimated to occur in between 1 in 5,600 and 1 in 26,550 newborns. However, this range may be an underestimate because not all medical professionals agree on the criteria for diagnosis of this condition, and because mild cases may never come to medical attention. For reasons that are unclear, the disorder occurs about 50 percent more often in males than in females.
Causes
It is unclear what genes are involved in craniofacial microsomia. This condition results from problems in the development of structures in the embryo called the first and second pharyngeal arches (also called branchial or visceral arches). Tissue layers in the six pairs of pharyngeal arches give rise to the muscles, arteries, nerves, and cartilage of the face and neck. Specifically, the first and second pharyngeal arches develop into the lower jaw, the nerves and muscles used for chewing and facial expression, the external ear, and the bones of the middle ear. Interference with the normal development of these structures can result in the abnormalities characteristic of craniofacial microsomia.
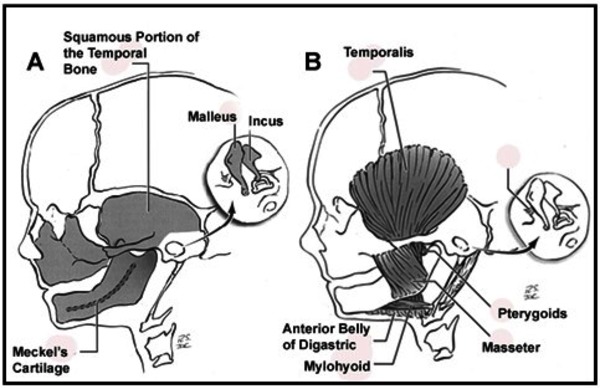
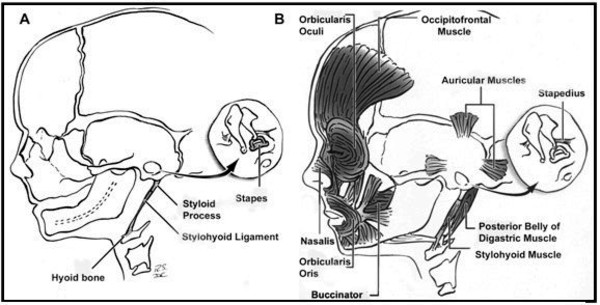
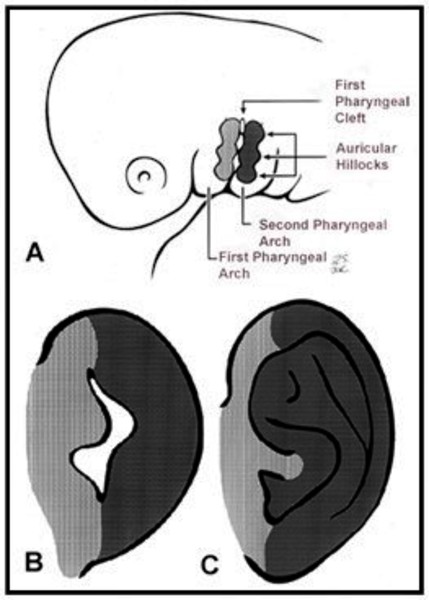
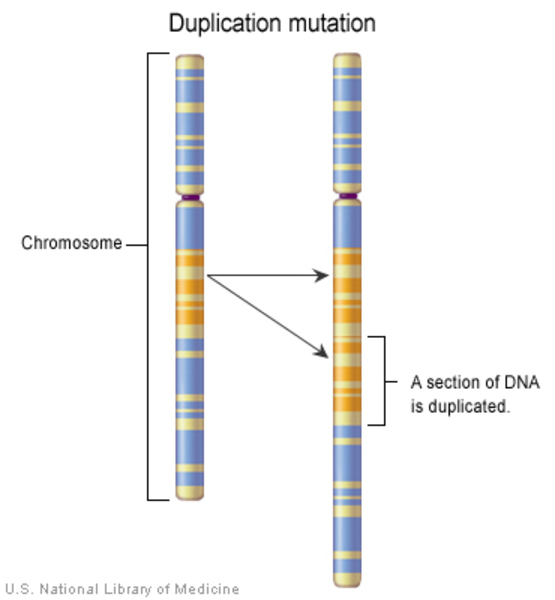
There are several factors that can disrupt the normal development of the first and second pharyngeal arches and lead to craniofacial microsomia. Some individuals with this condition have chromosomal abnormalities such as deletions or duplications of genetic material; these individuals often have additional developmental problems or malformations. Occasionally, craniofacial microsomia occurs in multiple members of a family in a pattern that suggests inheritance of a causative gene mutation, but the gene or genes involved are unknown. In other families, individuals seem to inherit a predisposition to the disorder. The risk of craniofacial microsomia can also be increased by environmental factors, such as certain drugs taken by the mother during pregnancy. In most affected individuals, the cause of the disorder is unknown.
It is not well understood why certain disruptions to development affect the first and second pharyngeal arches in particular. Researchers suggest that these structures may develop together in such a way that they respond as a unit to these disruptions.
Inheritance
Craniofacial microsomia most often occurs in a single individual in a family and is not inherited. If the condition is caused by a chromosomal abnormality, it may be inherited from one affected parent or it may result from a new abnormality in the chromosome and occur in people with no history of the disorder in their family.
In 1 to 2 percent of cases, craniofacial microsomia is inherited in an autosomal dominant pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder. In rare cases, the condition is inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. The gene or genes involved in craniofacial microsomia are unknown.


In some affected families, people seem to inherit an increased risk of developing craniofacial microsomia, not the condition itself. In these cases, some combination of genetic changes and environmental factors may be involved.
Other Names for This Condition
- Asymmetric hypoplasia of facial structures
- Auriculobranchiogenic dysplasia
- CFM
- Facioauriculovertebral dysplasia
- FAV
- First and second branchial arch syndrome
- First and second pharyngeal arch syndromes
- Goldenhar syndrome
- Goldenhar-Gorlin syndrome
- Hemifacial microsomia
- HFM
- Lateral facial dysplasia
- OAV complex
- OAVS
- Oculoauriculovertebral spectrum
- Oral-mandibular-auricular syndrome
- Otomandibular dysostosis
- Unilateral intrauterine facial necrosis
- Unilateral mandibulofacial dysostosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Gougoutas AJ, Singh DJ, Low DW, Bartlett SP. Hemifacial microsomia: clinical features and pictographic representations of the OMENS classification system. Plast Reconstr Surg. 2007 Dec;120(7):112e-113e. doi: 10.1097/01.prs.0000287383.35963.5e. Citation on PubMed
- Hartsfield JK. Review of the etiologic heterogeneity of the oculo-auriculo-vertebral spectrum (Hemifacial Microsomia). Orthod Craniofac Res. 2007 Aug;10(3):121-8. doi: 10.1111/j.1601-6343.2007.00391.x. Citation on PubMed
- Johnson JM, Moonis G, Green GE, Carmody R, Burbank HN. Syndromes of the first and second branchial arches, part 1: embryology and characteristic defects. AJNR Am J Neuroradiol. 2011 Jan;32(1):14-9. doi: 10.3174/ajnr.A2072. Epub 2010 Mar 18. Citation on PubMed
- Johnson JM, Moonis G, Green GE, Carmody R, Burbank HN. Syndromes of the first and second branchial arches, part 2: syndromes. AJNR Am J Neuroradiol. 2011 Feb;32(2):230-7. doi: 10.3174/ajnr.A2073. Epub 2010 Apr 1. Citation on PubMed
- Keogh IJ, Troulis MJ, Monroy AA, Eavey RD, Kaban LB. Isolated microtia as a marker for unsuspected hemifacial microsomia. Arch Otolaryngol Head Neck Surg. 2007 Oct;133(10):997-1001. doi: 10.1001/archotol.133.10.997. Citation on PubMed
- Rahbar R, Robson CD, Mulliken JB, Schwartz L, Dicanzio J, Kenna MA, McGill TJ, Healy GB. Craniofacial, temporal bone, and audiologic abnormalities in the spectrum of hemifacial microsomia. Arch Otolaryngol Head Neck Surg. 2001 Mar;127(3):265-71. doi: 10.1001/archotol.127.3.265. Citation on PubMed
- Tasse C, Bohringer S, Fischer S, Ludecke HJ, Albrecht B, Horn D, Janecke A, Kling R, Konig R, Lorenz B, Majewski F, Maeyens E, Meinecke P, Mitulla B, Mohr C, Preischl M, Umstadt H, Kohlhase J, Gillessen-Kaesbach G, Wieczorek D. Oculo-auriculo-vertebral spectrum (OAVS): clinical evaluation and severity scoring of 53 patients and proposal for a new classification. Eur J Med Genet. 2005 Oct-Dec;48(4):397-411. doi: 10.1016/j.ejmg.2005.04.015. Epub 2005 Jun 8. Citation on PubMed
- Vendramini-Pittoli S, Kokitsu-Nakata NM. Oculoauriculovertebral spectrum: report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clin Dysmorphol. 2009 Apr;18(2):67-77. doi: 10.1097/MCD.0b013e328323a7dd. Citation on PubMed
- Werler MM, Starr JR, Cloonan YK, Speltz ML. Hemifacial microsomia: from gestation to childhood. J Craniofac Surg. 2009 Mar;20 Suppl 1(Suppl 1):664-9. doi: 10.1097/SCS.0b013e318193d5d5. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.