

Description
Craniofacial-deafness-hand syndrome is characterized by distinctive facial features, profound hearing loss, and hand abnormalities.
The distinctive facial features of people with craniofacial-deafness-hand syndrome result from a variety of developmental abnormalities involving the skull (cranium) and face. Affected individuals often have underdeveloped or absent nasal bones resulting in a small nose, thin nostrils, and a flattened mid-face with a flat nasal bridge. Individuals with this condition typically also have widely spaced eyes (ocular hypertelorism), narrowed openings of the eyes (narrowed palpebral fissures), a small upper jaw (hypoplastic maxilla), and a small mouth with pursed lips.

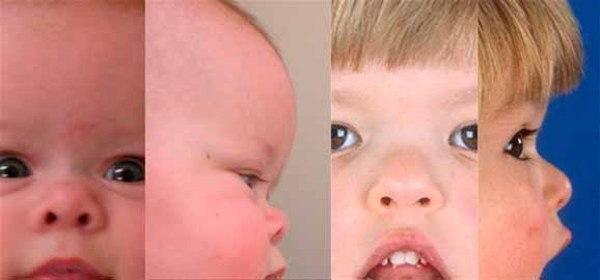


People with this condition also have profound hearing loss that is caused by abnormalities in the inner ear (sensorineural deafness). Hearing loss in these individuals is present from birth.

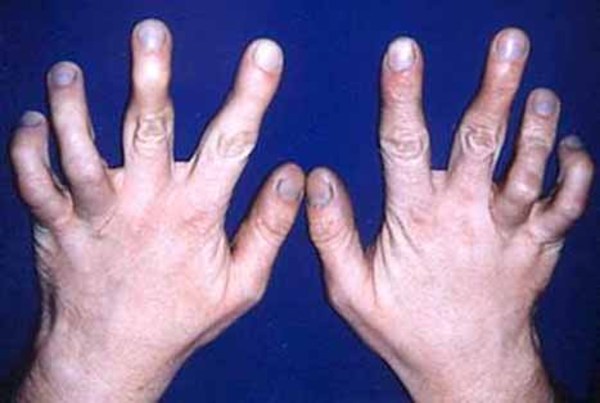
In affected individuals, a common abnormality of the muscles in the hand is a malformation in which all of the fingers are angled outward toward the fifth finger (ulnar deviation). People with craniofacial-deafness-hand syndrome may also have permanently bent third, fourth, and fifth fingers (camptodactyly), which can limit finger movement and lead to joint deformities called contractures. Contractures in the wrist can further impair hand movements.

Frequency
Craniofacial-deafness-hand syndrome is an extremely rare condition. Only a few cases have been reported in the scientific literature.
Causes
Craniofacial-deafness-hand syndrome is caused by mutations in the PAX3 gene. The PAX3 gene plays a critical role in the formation of tissues and organs during embryonic development. To perform this function, the gene provides instructions for making a protein that attaches (binds) to specific areas of DNA to help control the activity of particular genes. During embryonic development, the PAX3 gene is active in cells called neural crest cells. These cells migrate from the developing spinal cord to specific regions in the embryo. The protein produced from the PAX3 gene directs the activity of other genes that signal neural crest cells to form specialized tissues or cell types. These include some nerve tissues, bones in the face and skull (craniofacial bones), and muscle tissue.
At least one PAX3 gene mutation has been identified in individuals with craniofacial-deafness-hand syndrome. This mutation appears to affect the ability of the PAX3 protein to bind to DNA. As a result, the PAX3 protein cannot control the activity of other genes and cannot regulate the differentiation of neural crest cells. A lack of specialization of neural crest cells leads to the impaired growth of craniofacial bones, nerve tissue, and muscles seen in craniofacial-deafness-hand syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- CDHS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Asher JH Jr, Sommer A, Morell R, Friedman TB. Missense mutation in the paired domain of PAX3 causes craniofacial-deafness-hand syndrome. Hum Mutat. 1996;7(1):30-5. doi: 10.1002/(SICI)1098-1004(1996)7:13.0.CO;2-T. Citation on PubMed
- Birrane G, Soni A, Ladias JA. Structural basis for DNA recognition by the human PAX3 homeodomain. Biochemistry. 2009 Feb 17;48(6):1148-55. doi: 10.1021/bi802052y. Citation on PubMed
- Sommer A, Bartholomew DW. Craniofacial-deafness-hand syndrome revisited. Am J Med Genet A. 2003 Nov 15;123A(1):91-4. doi: 10.1002/ajmg.a.20501. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.