

Description
Cornelia de Lange syndrome is a developmental disorder that affects many parts of the body. The features of this disorder vary widely among affected individuals and range from relatively mild to severe.
Cornelia de Lange syndrome is characterized by slow growth before and after birth leading to short stature; intellectual disability that is usually moderate to severe; and abnormalities of bones in the arms, hands, and fingers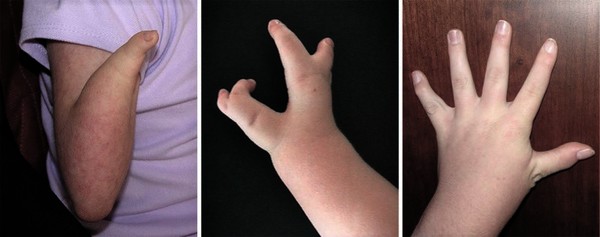 . Most people with Cornelia de Lange syndrome also have distinctive facial features, including arched eyebrows that often meet in the middle (synophrys
. Most people with Cornelia de Lange syndrome also have distinctive facial features, including arched eyebrows that often meet in the middle (synophrys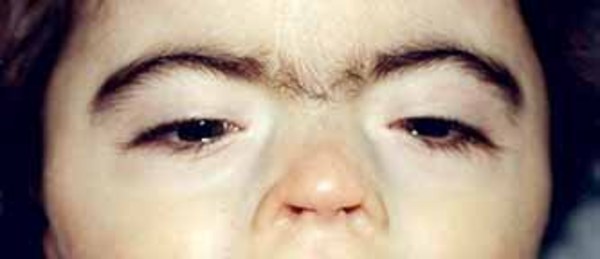 ), long eyelashes
), long eyelashes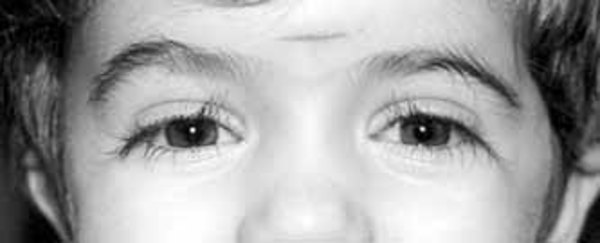 , low-set ears, small and widely spaced teeth, and a small and upturned nose. Many affected individuals also have features similar to autism spectrum disorder, a developmental condition that affects communication and social interaction.
, low-set ears, small and widely spaced teeth, and a small and upturned nose. Many affected individuals also have features similar to autism spectrum disorder, a developmental condition that affects communication and social interaction.
Additional signs and symptoms of Cornelia de Lange syndrome can include excessive body hair (hypertrichosis), an unusually small head (microcephaly ), hearing loss, and problems with the digestive tract
), hearing loss, and problems with the digestive tract . Some people with this condition are born with an opening in the roof of the mouth called a cleft palate
. Some people with this condition are born with an opening in the roof of the mouth called a cleft palate . Seizures, heart defects, and eye problems have also been reported in people with this condition.
. Seizures, heart defects, and eye problems have also been reported in people with this condition.
Frequency
Although the exact incidence is unknown, Cornelia de Lange syndrome likely affects 1 in 10,000 to 30,000 newborns. The condition is probably underdiagnosed because affected individuals with mild or uncommon features may never be recognized as having Cornelia de Lange syndrome.
Causes
Cornelia de Lange syndrome can result from variants (also called mutations) in one of several genes. Variants in the NIPBL gene have been identified in more than half of all people with this condition. Variants in other genes, including SMC1A, HDAC8, RAD21, SMC3, and others, are much less common.
The proteins produced from most of the genes involved in Cornelia de Lange syndrome contribute to the structure or function of the cohesin complex, a group of proteins with an important role in directing development before birth. Within cells, the cohesin complex helps regulate the structure and organization of chromosomes, stabilize cells' genetic information, and repair damaged DNA. The cohesin complex also regulates the activity of certain genes that guide the development of the face, limbs, and other parts of the body. Other genes that are involved in rare cases of Cornelia de Lange syndrome also control gene activity and are likely important during development.
Variants in the NIPBL, SMC1A, HDAC8, RAD21, and SMC3 genes cause Cornelia de Lange syndrome by impairing the function of the cohesin complex, which disrupts gene regulation during critical stages of early development. Variants in other genes are also thought to disrupt the regulation of genes important for development.
The features of Cornelia de Lange syndrome vary widely, and the severity of the disorder can differ even in individuals with the same gene variant. Researchers suspect that additional genetic or environmental factors may be important for determining the specific signs and symptoms in each individual. In general, SMC1A, RAD21, and SMC3 gene variants cause milder signs and symptoms than NIPBL gene variants. Variants in the HDAC8 gene cause a somewhat different set of features, including delayed closure of the "soft spot" on the head (the anterior fontanelle) in infancy, widely spaced eyes , and dental abnormalities. Like affected individuals with NIPBL gene variants, those with HDAC8 gene variants may have significant intellectual disability.
, and dental abnormalities. Like affected individuals with NIPBL gene variants, those with HDAC8 gene variants may have significant intellectual disability.
In about 15 percent of cases, the cause of Cornelia de Lange syndrome is unknown. Researchers are looking for additional changes in the known genes, as well as variants in other genes, that may cause this condition.
Inheritance
When Cornelia de Lange syndrome is caused by variants in the NIPBL, RAD21, or SMC3 gene, the condition is considered to have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new gene variants and occur in people with no history of the condition in their family.
of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new gene variants and occur in people with no history of the condition in their family.
When Cornelia de Lange syndrome is caused by variants in the HDAC8 or SMC1A gene, the condition has an X-linked dominant pattern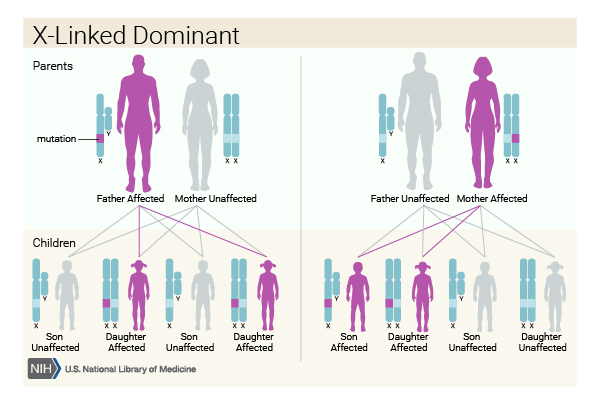 of inheritance. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes
of inheritance. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes . Studies of X-linked Cornelia de Lange syndrome indicate that one copy of the altered gene in each cell may be sufficient to cause the condition. Affected females, who have two X chromosomes, may have milder signs and symptoms than affected males, who have only one X chromosome. Most X-linked cases result from new variants in the HDAC8 or SMC1A gene and occur in people with no history of the condition in their family.
. Studies of X-linked Cornelia de Lange syndrome indicate that one copy of the altered gene in each cell may be sufficient to cause the condition. Affected females, who have two X chromosomes, may have milder signs and symptoms than affected males, who have only one X chromosome. Most X-linked cases result from new variants in the HDAC8 or SMC1A gene and occur in people with no history of the condition in their family.
Other Names for This Condition
- BDLS
- Brachmann-de Lange syndrome
- CdLS
- De Lange syndrome
- Typus degenerativus amstelodamensis
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Boyle MI, Jespersgaard C, Brondum-Nielsen K, Bisgaard AM, Tumer Z. Cornelia de Lange syndrome. Clin Genet. 2015 Jul;88(1):1-12. doi: 10.1111/cge.12499. Epub 2014 Oct 28. Citation on PubMed
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012 Sep 13;489(7415):313-7. doi: 10.1038/nature11316. Citation on PubMed or Free article on PubMed Central
- Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange Syndrome. 2005 Sep 16 [updated 2020 Oct 15]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1104/ Citation on PubMed
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Monnich M, Yan Y, Xu W, Gil-Rodriguez MC, Clark D, Hakonarson H, Halbach S, Michelis LD, Rampuria A, Rossier E, Spranger S, Van Maldergem L, Lynch SA, Gillessen-Kaesbach G, Ludecke HJ, Ramsay RG, McKay MJ, Krantz ID, Xu H, Horsfield JA, Kaiser FJ. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. 2012 Jun 8;90(6):1014-27. doi: 10.1016/j.ajhg.2012.04.019. Epub 2012 May 24. Citation on PubMed or Free article on PubMed Central
- Kaiser FJ, Ansari M, Braunholz D, Concepcion Gil-Rodriguez M, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillen-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I; Care4Rare Canada Consortium; Mohammed S, Moran E, Mortier GR, Moser JA, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Perez-Aytes A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC; University of Washington Center for Mendelian Genomics; Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pie J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014 Jun 1;23(11):2888-900. doi: 10.1093/hmg/ddu002. Epub 2014 Jan 8. Citation on PubMed or Free article on PubMed Central
- Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, Ishman SL, Kerr LM, Levin AV, Mulder PA, Ramos FJ, Wierzba J, Ajmone PF, Axtell D, Blagowidow N, Cereda A, Costantino A, Cormier-Daire V, FitzPatrick D, Grados M, Groves L, Guthrie W, Huisman S, Kaiser FJ, Koekkoek G, Levis M, Mariani M, McCleery JP, Menke LA, Metrena A, O'Connor J, Oliver C, Pie J, Piening S, Potter CJ, Quaglio AL, Redeker E, Richman D, Rigamonti C, Shi A, Tumer Z, Van Balkom IDC, Hennekam RC. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018 Oct;19(10):649-666. doi: 10.1038/s41576-018-0031-0. Citation on PubMed
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004 Jun;36(6):631-5. doi: 10.1038/ng1364. Epub 2004 May 16. Citation on PubMed or Free article on PubMed Central
- Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum Mutat. 2013 Dec;34(12):1589-96. doi: 10.1002/humu.22430. Epub 2013 Sep 16. Citation on PubMed or Free article on PubMed Central
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006 May;38(5):528-30. doi: 10.1038/ng1779. Epub 2006 Apr 9. Citation on PubMed
- Olley G, Ansari M, Bengani H, Grimes GR, Rhodes J, von Kriegsheim A, Blatnik A, Stewart FJ, Wakeling E, Carroll N, Ross A, Park SM; Deciphering Developmental Disorders Study; Bickmore WA, Pradeepa MM, FitzPatrick DR. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nat Genet. 2018 Mar;50(3):329-332. doi: 10.1038/s41588-018-0042-y. Epub 2018 Jan 29. Citation on PubMed
- Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, Watrin E, Wendt KS, Cereda A, Cittaro D, Gillessen-Kaesbach G, Lazarevic D, Mariani M, Russo S, Werner R, Krawitz P, Larizza L, Selicorni A, Kaiser FJ. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin Genet. 2016 Jan;89(1):74-81. doi: 10.1111/cge.12564. Epub 2015 Feb 25. Citation on PubMed
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004 Jun;36(6):636-41. doi: 10.1038/ng1363. Epub 2004 May 16. Citation on PubMed
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, Tos T, Koparir A, Koparir E, Beck CR, Gu S, Aslan H, Yuregir OO, Al Rubeaan K, Alnaqeb D, Alshammari MJ, Bayram Y, Atik MM, Aydin H, Geckinli BB, Seven M, Ulucan H, Fenercioglu E, Ozen M, Jhangiani S, Muzny DM, Boerwinkle E, Tuysuz B, Alkuraya FS, Gibbs RA, Lupski JR. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015 Feb;125(2):636-51. doi: 10.1172/JCI77435. Epub 2015 Jan 9. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.