

Description
CLN11 disease is a disorder that primarily affects the nervous system. Individuals with this condition typically show signs and symptoms in adolescence or early adulthood. This condition is characterized by recurrent seizures (epilepsy), vision loss, problems with balance and coordination (cerebellar ataxia), and a decline in intellectual function.
Seizures in CLN11 disease often involve a loss of consciousness, muscle stiffness (rigidity), and generalized convulsions (tonic-clonic seizures).
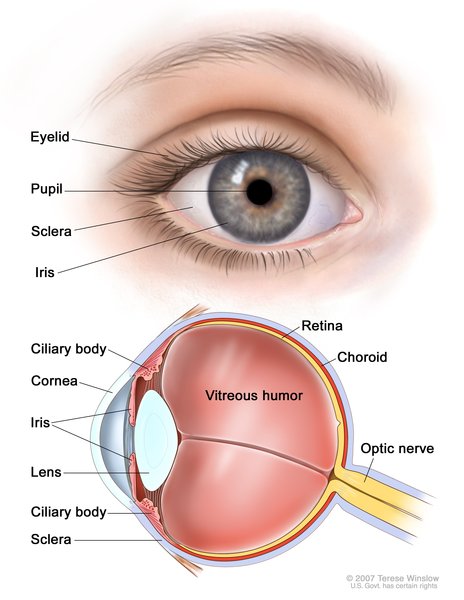
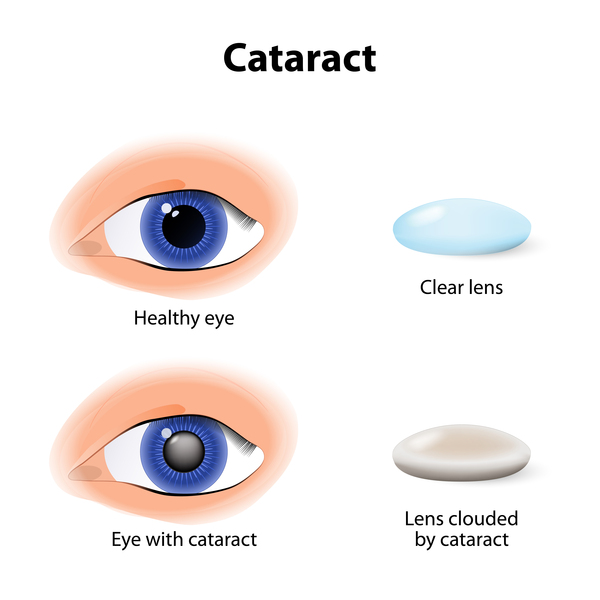
Vision loss is gradual over time and is due to a condition called retinitis pigmentosa, which is caused by the breakdown of the light-sensitive layer at the back of the eye (retina). People with CLN11 disease can also develop clouding of the lenses of the eyes (cataracts) and rapid, involuntary eye movements (nystagmus).
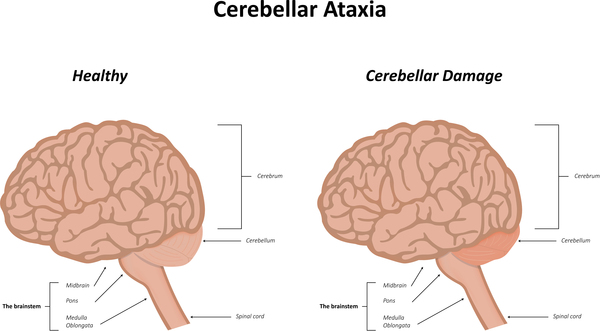
Affected individuals can also develop muscle twitches (myoclonus), walking problems and falling (gait disturbance), and impaired speech (dysarthria). Over time, people with CLN11 disease develop short-term memory loss and loss of executive function, which is the ability to plan and implement problem-solving strategies and actions. They may also become irritable and impulsive. Some affected individuals experience visual hallucinations involving people or animals.
CLN11 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs). All of these disorders affect the nervous system and typically cause progressive problems with vision, movement, and thinking ability. The different NCLs are distinguished by their genetic cause. Each disease type is given the designation "CLN," meaning ceroid lipofuscinosis, neuronal, and then a number to indicate its subtype.
Frequency
The prevalence of CLN11 disease is unknown; at least 11 cases have been described in the scientific literature.
Causes
CLN11 disease results from mutations in the GRN gene. This gene provides instructions for making a protein called progranulin. Progranulin is active in many different tissues in the body, where it helps control the growth, division, and survival of cells. Progranulin's function in the brain is not well understood, although it appears to play an important role in the survival of nerve cells (neurons).

GRN gene mutations that cause CLN11 disease result in a complete loss of functional progranulin protein. This lack of progranulin causes the death of nerve cells in the brain, although the exact mechanism is unknown. Widespread loss of neurons in CLN11 disease leads to the development of signs and symptoms in adolescence or early adulthood.
Inheritance
CLN11 disease is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations. Having a mutation in both copies of the GRN gene eliminates production of any functional progranulin protein.
, which means both copies of the gene in each cell have mutations. Having a mutation in both copies of the GRN gene eliminates production of any functional progranulin protein.
The parents of individuals with CLN11 disease each carry one copy of the mutated GRN gene in every cell and generally produce about half the normal amount of progranulin protein. Individuals with one GRN gene mutation typically do not show signs and symptoms of CLN11 disease, but they may develop another condition called GRN-related frontotemporal lobar degeneration in which cognitive decline begins between a person's forties and sixties. Some people with two GRN gene mutations that allow the production of some functional progranulin protein develop GRN-related frontotemporal lobar degeneration.
Other Names for This Condition
- Ceroid lipofuscinosis, neuronal, 11
- GRN-related neuronal ceroid-lipofuscinosis
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. 2016 May;41:200.e1-200.e5. doi: 10.1016/j.neurobiolaging.2016.02.019. Epub 2016 Mar 3. Citation on PubMed
- Canafoglia L, Morbin M, Scaioli V, Pareyson D, D'Incerti L, Fugnanesi V, Tagliavini F, Berkovic SF, Franceschetti S. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia. 2014 Jun;55(6):e56-9. doi: 10.1111/epi.12632. Epub 2014 Apr 29. Citation on PubMed
- Faber I, Prota JR, Martinez AR, Lopes-Cendes I, Franca MC Junior. A new phenotype associated with homozygous GRN mutations: complicated spastic paraplegia. Eur J Neurol. 2017 Jan;24(1):e3-e4. doi: 10.1111/ene.13194. No abstract available. Citation on PubMed
- Huin V, Barbier M, Bottani A, Lobrinus JA, Clot F, Lamari F, Chat L, Rucheton B, Fluchere F, Auvin S, Myers P, Gelot A, Camuzat A, Caillaud C, Jornea L, Forlani S, Saracino D, Duyckaerts C, Brice A, Durr A, Le Ber I. Homozygous GRN mutations: new phenotypes and new insights into pathological and molecular mechanisms. Brain. 2020 Jan 1;143(1):303-319. doi: 10.1093/brain/awz377. Citation on PubMed
- Kamate M, Detroja M, Hattiholi V. Neuronal ceroid lipofuscinosis type-11 in an adolescent. Brain Dev. 2019 Jun;41(6):542-545. doi: 10.1016/j.braindev.2019.03.004. Epub 2019 Mar 25. Citation on PubMed
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012 Jun 8;90(6):1102-7. doi: 10.1016/j.ajhg.2012.04.021. Epub 2012 May 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.