

Description
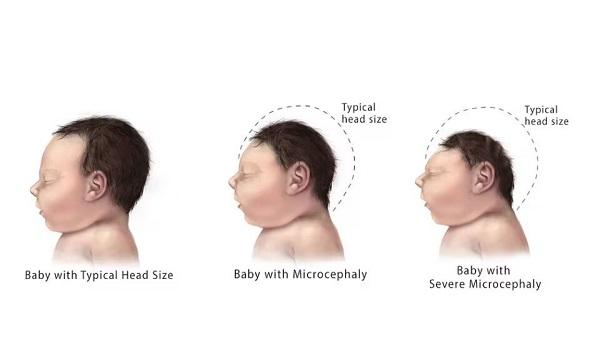
CLN1 disease is an inherited disorder that primarily affects the nervous system. Individuals with this condition have normal development in infancy, but typically by 18 months they become increasingly irritable and begin to lose previously acquired skills (developmental regression). In affected children, nerve cells in the brain die over time, leading to an overall loss of brain tissue (brain atrophy) and an unusually small head (microcephaly). Children with CLN1 disease have decreased muscle tone (hypotonia), intellectual and motor disability, and rarely are able to speak or walk. Some affected children develop repetitive hand movements. By age 2, individuals with this condition often have muscle twitches (myoclonus), recurrent seizures (epilepsy), and vision loss. Some affected children develop frequent respiratory infections. As the condition worsens, children have severe feeding difficulties that often require a feeding tube. Children with CLN1 disease usually do not survive past childhood.

Some people with CLN1 disease do not develop symptoms until later in childhood or in adulthood. As with younger affected children, older individuals develop a decline in intellectual function, myoclonus, epilepsy, and vision loss. In these individuals, life expectancy depends on when signs and symptoms of CLN1 disease develop and their severity; affected individuals may survive only into adolescence or through adulthood. Adults with CLN1 disease may also have movement disorders, including impaired muscle coordination (ataxia) or a pattern of movement abnormalities known as parkinsonism.
CLN1 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs), which may also be collectively referred to as Batten disease. All these disorders affect the nervous system and typically cause worsening problems with vision, movement, and thinking ability. The different NCLs are distinguished by their genetic cause. Each disease type is given the designation "CLN," meaning ceroid lipofuscinosis, neuronal, and then a number to indicate its subtype.
Frequency
The incidence of CLN1 disease is unknown; more than 200 cases have been described in the scientific literature. Collectively, all forms of NCL affect an estimated 1 in 100,000 individuals worldwide. NCLs are more common in Finland, where approximately 1 in 12,500 individuals are affected.
Causes
Mutations in the PPT1 gene cause CLN1 disease. The PPT1 gene provides instructions for making an enzyme called palmitoyl-protein thioesterase 1. This enzyme is active in cell compartments called lysosomes, which digest and recycle different types of molecules. Palmitoyl-protein thioesterase 1 removes fats called long-chain fatty acids from certain proteins, which helps to break down the proteins. Palmitoyl-protein thioesterase 1 is also thought to be involved in a variety of other cell functions.

PPT1 gene mutations that cause CLN1 disease decrease or eliminate the production or function of palmitoyl-protein thioesterase 1. A reduction of functional enzyme impairs the removal of fatty acids from certain proteins. These partially broken down fats and proteins accumulate in lysosomes. While accumulation of these substances occurs in cells throughout the body, nerve cells appear to be particularly vulnerable to damage caused by the abnormal cell materials. Early and widespread loss of nerve cells in CLN1 disease leads to severe signs and symptoms and death in childhood.
In the later-onset cases of CLN1 disease, PPT1 gene mutations result in the production of a palmitoyl-protein thioesterase 1 enzyme that has a reduced level of normal function; however, protein function in these individuals is higher than in those who have the condition beginning in early childhood. As a result, long-chain fatty acids are removed from some proteins, allowing for a small amount of proteins to be broken down. Since it takes longer for these substances to accumulate in lysosomes and cause nerve cell death, the signs and symptoms of CLN1 disease in these individuals occur later in life.

Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- CLN1
- Infantile Batten disease
- Infantile neuronal ceroid lipofuscinosis
- Neuronal ceroid lipofuscinosis 1
- Neuronal ceroid lipofuscinosis, infantile
- Santavuori-Haltia disease
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Getty AL, Pearce DA. Interactions of the proteins of neuronal ceroid lipofuscinosis: clues to function. Cell Mol Life Sci. 2011 Feb;68(3):453-74. doi: 10.1007/s00018-010-0468-6. Epub 2010 Aug 1. Citation on PubMed or Free article on PubMed Central
- Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009 Apr;1793(4):697-709. doi: 10.1016/j.bbamcr.2008.11.004. Epub 2008 Nov 24. Citation on PubMed
- Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, Mukherjee AB. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest. 2008 Sep;118(9):3075-86. doi: 10.1172/JCI33482. Citation on PubMed or Free article on PubMed Central
- Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta. 2013 Nov;1832(11):1866-81. doi: 10.1016/j.bbadis.2013.01.019. Epub 2013 Feb 9. Citation on PubMed
- Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012 Jan;33(1):42-63. doi: 10.1002/humu.21624. Epub 2011 Nov 16. Citation on PubMed
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012 Jul 10;79(2):183-91. doi: 10.1212/WNL.0b013e31825f0547. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.