

Description
Cleidocranial dysplasia is a condition that primarily affects development of the bones and teeth. Signs and symptoms of cleidocranial dysplasia can vary widely in severity, even within the same family.
Individuals with cleidocranial dysplasia usually have underdeveloped or absent collarbones, also called clavicles ("cleido-" in the condition name refers to these bones). As a result, their shoulders are narrow and sloping, can be brought unusually close together in front of the body, and in some cases can be made to meet in the middle of the body. Delayed maturation of the skull (cranium) is also characteristic of this condition, including delayed closing of the growth lines where the bones of the skull meet (sutures) and larger than normal spaces (fontanelles) between the skull bones that are noticeable as "soft spots" on the heads of infants. The fontanelles normally close in early childhood, but they may remain open throughout life in people with this disorder. Some individuals with cleidocranial dysplasia have extra pieces of bone called Wormian bones within the sutures.
Affected individuals are often shorter than other members of their family at the same age. Many also have short, tapered fingers and broad thumbs; flat feet; knock knees; short shoulder blades (scapulae); and an abnormal curvature of the spine (scoliosis). Typical facial features include a wide, short skull (brachycephaly); a prominent forehead; wide-set eyes (hypertelorism); a flat nose; and a small upper jaw.

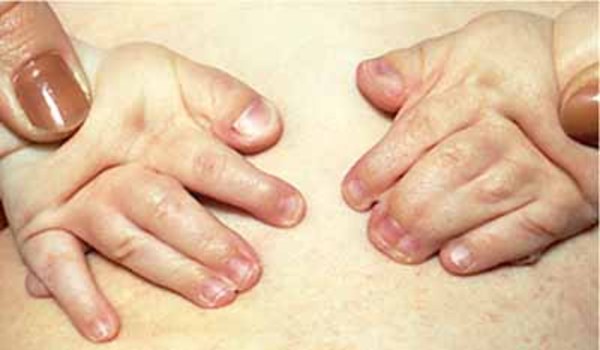

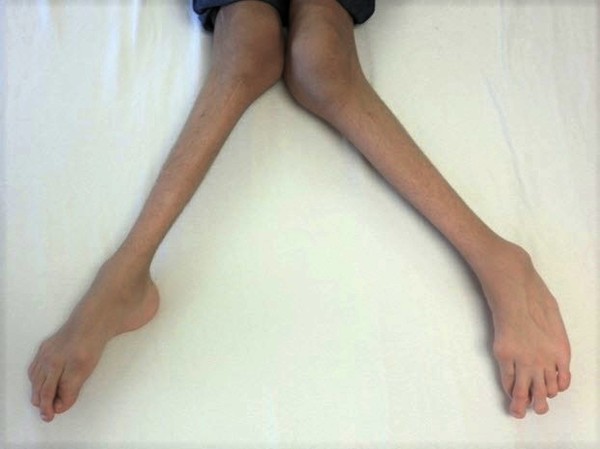


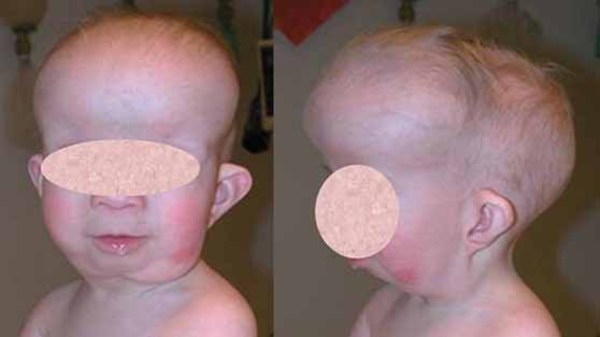

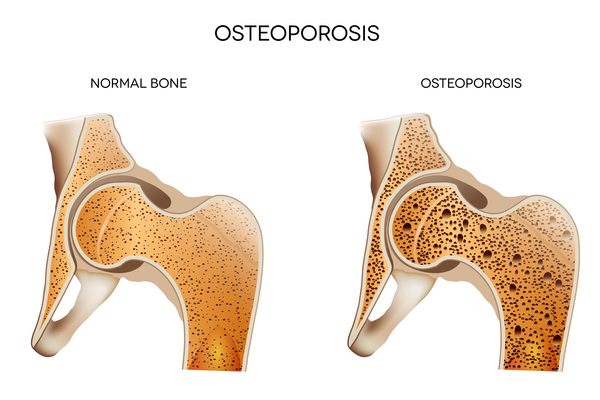
Individuals with cleidocranial dysplasia often have decreased bone density (osteopenia) and may develop osteoporosis, a condition that makes bones progressively more brittle and prone to fracture, at a relatively early age. Women with cleidocranial dysplasia have an increased risk of requiring a cesarean section when delivering a baby, due to a narrow pelvis preventing passage of the infant's head.
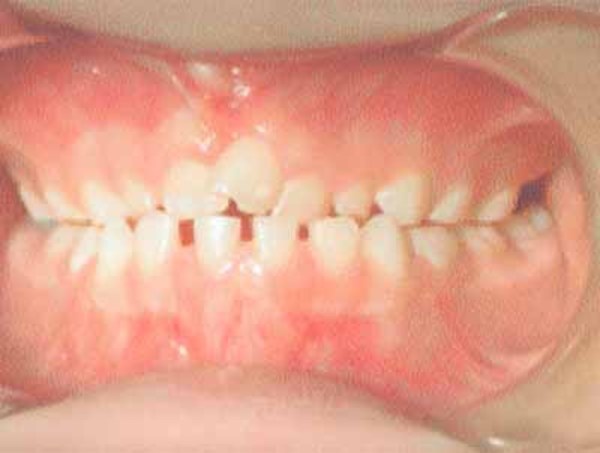
Dental abnormalities are very common in cleidocranial dysplasia and can include delayed loss of the primary (baby) teeth; delayed appearance of the secondary (adult) teeth; unusually shaped, peg-like teeth; misalignment of the teeth and jaws (malocclusion); and extra teeth, sometimes accompanied by cysts in the gums.
In addition to skeletal and dental abnormalities, people with cleidocranial dysplasia may have hearing loss and are prone to sinus and ear infections. Some young children with this condition are mildly delayed in the development of motor skills such as crawling and walking, but intelligence is unaffected.
Frequency
Cleidocranial dysplasia occurs in approximately 1 per million individuals worldwide. It is likely underdiagnosed because many affected individuals have mild signs and symptoms.
Causes
Cleidocranial dysplasia is usually caused by mutations in the RUNX2 gene. This gene provides instructions for making a protein that is involved in the development and maintenance of teeth, bones, and cartilage. Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone (a process called ossification), except for the cartilage that continues to cover and protect the ends of bones and is present in the nose, airways, and external ears.
The RUNX2 protein is a transcription factor, which means it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. Researchers believe that the RUNX2 protein acts as a "master switch," regulating a number of other genes involved in the development of cells that build bones (osteoblasts) and in the development of teeth.
The RUNX2 gene mutations that cause cleidocranial dysplasia reduce or eliminate the activity of the protein produced from one copy of the RUNX2 gene in each cell, decreasing the total amount of functional RUNX2 protein. This shortage of functional RUNX2 protein interferes with the normal development of bones, cartilage, and teeth, resulting in the signs and symptoms of cleidocranial dysplasia. In rare cases, individuals with a deletion of genetic material that includes RUNX2 and other nearby genes may experience additional features, such as developmental delay, resulting from the loss of these genes.
In about 30 percent of individuals with cleidocranial dysplasia, no mutation in the RUNX2 gene has been found. The cause of the condition in these individuals is unknown.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Some affected individuals inherit the mutation from one affected parent. Often the parent is mildly affected, and in some cases had not previously been recognized as having the disorder. Other cases result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- Cleidocranial dysostosis
- Dento-osseous dysplasia
- Marie-Sainton syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bruderer M, Richards RG, Alini M, Stoddart MJ. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. 2014 Oct 23;28:269-86. doi: 10.22203/ecm.v028a19. Citation on PubMed
- Bufalino A, Paranaiba LM, Gouvea AF, Gueiros LA, Martelli-Junior H, Junior JJ, Lopes MA, Graner E, De Almeida OP, Vargas PA, Coletta RD. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis. 2012 Mar;18(2):184-90. doi: 10.1111/j.1601-0825.2011.01862.x. Epub 2011 Oct 24. Citation on PubMed
- Cohen MM Jr. Biology of RUNX2 and Cleidocranial Dysplasia. J Craniofac Surg. 2013 Jan;24(1):130-3. doi: 10.1097/SCS.0b013e3182636b7e. Citation on PubMed
- D'Alessandro G, Tagariello T, Piana G. Cleidocranial dysplasia: etiology and stomatognathic and craniofacial abnormalities. Minerva Stomatol. 2010 Mar;59(3):117-27. English, Italian. Citation on PubMed
- Dincsoy Bir F, Dinckan N, Guven Y, Bas F, Altunoglu U, Kuvvetli SS, Poyrazoglu S, Toksoy G, Kayserili H, Uyguner ZO. Cleidocranial dysplasia: Clinical, endocrinologic and molecular findings in 15 patients from 11 families. Eur J Med Genet. 2017 Mar;60(3):163-168. doi: 10.1016/j.ejmg.2016.12.007. Epub 2016 Dec 24. Citation on PubMed
- Jaruga A, Hordyjewska E, Kandzierski G, Tylzanowski P. Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlation. Clin Genet. 2016 Nov;90(5):393-402. doi: 10.1111/cge.12812. Epub 2016 Jun 30. Citation on PubMed
- Machol K, Mendoza-Londono R, Lee B. Cleidocranial Dysplasia Spectrum Disorder. 2006 Jan 3 [updated 2023 Apr 13]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1513/ Citation on PubMed
- Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the South African experience. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013 Jan;115(1):46-55. doi: 10.1016/j.oooo.2012.07.435. Epub 2012 Oct 24. Citation on PubMed
- Ryoo HM, Wang XP. Control of tooth morphogenesis by Runx2. Crit Rev Eukaryot Gene Expr. 2006;16(2):143-54. doi: 10.1615/critreveukargeneexpr.v16.i2.30. Citation on PubMed
- Stevenson DA, Carey JC, Byrne JL, Srisukhumbowornchai S, Feldkamp ML. Analysis of skeletal dysplasias in the Utah population. Am J Med Genet A. 2012 May;158A(5):1046-54. doi: 10.1002/ajmg.a.35327. Epub 2012 Mar 27. Citation on PubMed
- Vimalraj S, Arumugam B, Miranda PJ, Selvamurugan N. Runx2: Structure, function, and phosphorylation in osteoblast differentiation. Int J Biol Macromol. 2015;78:202-8. doi: 10.1016/j.ijbiomac.2015.04.008. Epub 2015 Apr 13. Citation on PubMed
- Wysokinski D, Pawlowska E, Blasiak J. RUNX2: A Master Bone Growth Regulator That May Be Involved in the DNA Damage Response. DNA Cell Biol. 2015 May;34(5):305-15. doi: 10.1089/dna.2014.2688. Epub 2015 Jan 2. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.