

Description
CHD2 myoclonic encephalopathy is a condition characterized by recurrent seizures (epilepsy), abnormal brain function (encephalopathy), and intellectual disability. Epilepsy begins in childhood, typically between ages 6 months and 4 years. Each individual may experience a variety of seizure types. The most common are myoclonic seizures, which involve involuntary muscle twitches. Other seizure types include sudden episodes of weak muscle tone (atonic seizures); partial or complete loss of consciousness (absence seizures); seizures brought on by high body temperature (febrile seizure); or tonic-clonic seizures, which involve loss of consciousness, muscle rigidity, and convulsions. Some people with CHD2 myoclonic encephalopathy have photosensitive epilepsy, in which seizures are triggered by flashing lights. Some people with CHD2 myoclonic encephalopathy experience a type of seizure called atonic-myoclonic-absence seizure, which begins with a drop of the head, followed by loss of consciousness, then rigid movements of the arms. Epilepsy can worsen, causing prolonged episodes of seizure activity that last several minutes, known as status epilepticus. The seizures associated with CHD2 myoclonic encephalopathy are called refractory because they usually do not respond to therapy with anti-epileptic medications.
Other signs and symptoms of CHD2 myoclonic encephalopathy include intellectual disability that ranges from mild to severe and delayed development of speech. Rarely, individuals can have a loss of acquired skills (developmental regression) following the onset of epilepsy. Some people with CHD2 myoclonic encephalopathy have autism spectrum disorders, which are conditions characterized by impaired communication and social interaction. In some instances, areas with a loss of brain tissue (atrophy) have been found with medical imaging.
Frequency
The prevalence of CHD2 myoclonic encephalopathy is unknown; at least 32 cases have been described in the scientific literature.
Causes
As its name suggests, CHD2 myoclonic encephalopathy is caused by mutations in the CHD2 gene. This gene provides instructions for making a protein called chromodomain DNA helicase protein 2. This protein is found in cells throughout the body and regulates gene activity (expression) through a process known as chromatin remodeling. Chromatin is the complex of DNA and proteins that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. The role of chromodomain DNA helicase protein 2 in the brain is unknown. Researchers suspect that the protein may be involved in regulating the development and functioning of nerve cells.
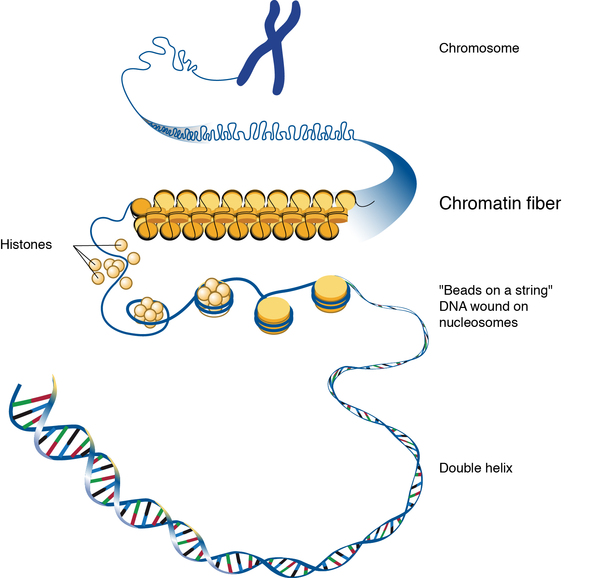
CHD2 gene mutations either prevent the production of any chromodomain DNA helicase protein 2 or lead to the production of a nonfunctional version of the protein. As a result, chromatin remodeling and gene expression normally regulated by chromodomain DNA helicase protein 2 are disrupted. It is unclear why CHD2 gene mutations seem to only affect nerve cells in the brain or how they lead to the signs and symptoms of CHD2 myoclonic encephalopathy.
Inheritance
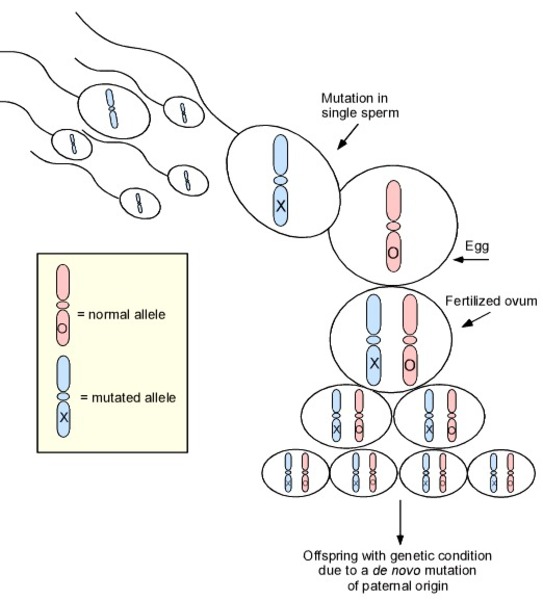
This condition is considered autosomal dominant, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

This condition results from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- CHD2 encephalopathy
- CHD2-related neurodevelopmental disorders
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Lund C, Brodtkorb E, Oye AM, Rosby O, Selmer KK. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav. 2014 Apr;33:18-21. doi: 10.1016/j.yebeh.2014.02.005. Epub 2014 Mar 12. Citation on PubMed
- Suls A, Jaehn JA, Kecskes A, Weber Y, Weckhuysen S, Craiu DC, Siekierska A, Djemie T, Afrikanova T, Gormley P, von Spiczak S, Kluger G, Iliescu CM, Talvik T, Talvik I, Meral C, Caglayan HS, Giraldez BG, Serratosa J, Lemke JR, Hoffman-Zacharska D, Szczepanik E, Barisic N, Komarek V, Hjalgrim H, Moller RS, Linnankivi T, Dimova P, Striano P, Zara F, Marini C, Guerrini R, Depienne C, Baulac S, Kuhlenbaumer G, Crawford AD, Lehesjoki AE, de Witte PA, Palotie A, Lerche H, Esguerra CV, De Jonghe P, Helbig I; EuroEPINOMICS RES Consortium. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013 Nov 7;93(5):967-75. doi: 10.1016/j.ajhg.2013.09.017. Epub 2013 Oct 24. Citation on PubMed or Free article on PubMed Central
- Thomas RH, Zhang LM, Carvill GL, Archer JS, Heavin SB, Mandelstam SA, Craiu D, Berkovic SF, Gill DS, Mefford HC, Scheffer IE; EuroEPINOMICS RES Consortium. CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology. 2015 Mar 3;84(9):951-8. doi: 10.1212/WNL.0000000000001305. Epub 2015 Feb 11. Citation on PubMed or Free article on PubMed Central
- Trivisano M, Striano P, Sartorelli J, Giordano L, Traverso M, Accorsi P, Cappelletti S, Claps DJ, Vigevano F, Zara F, Specchio N. CHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures. Epilepsy Behav. 2015 Oct;51:53-6. doi: 10.1016/j.yebeh.2015.06.029. Epub 2015 Aug 7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.