

Description
Camurati-Engelmann disease is a skeletal condition that is characterized by abnormally thick bones (hyperostosis) in the arms, legs, and skull.
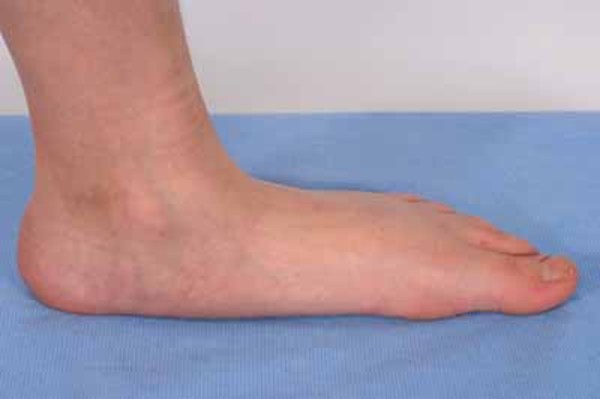
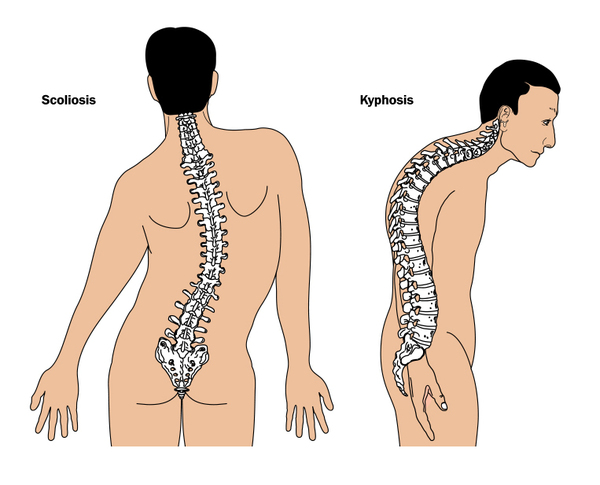
The thick limb bones can lead to bone pain and muscle weakness in the arms and legs and cause individuals with Camurati-Engelmann disease to tire quickly. Bone pain ranges from mild to severe and can increase with stress, activity, or cold weather. Leg weakness can make it difficult to stand up from a seated position and some affected individuals develop a waddling or unsteady walk. Additional limb abnormalities include joint deformities (contractures), knock knees, and flat feet (pes planus). Swelling and redness (erythema) of the limbs and an abnormal curvature of the spine can also occur.
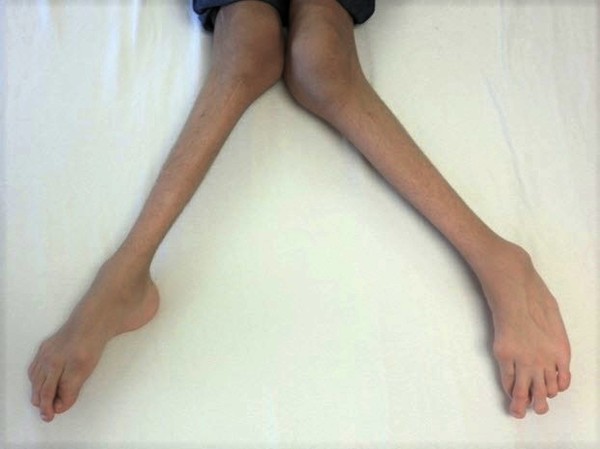
Individuals with Camurati-Engelmann disease may have an unusually thick skull, which can lead to an abnormally large head (macrocephaly) and lower jaw (mandible), a prominent forehead (frontal bossing), and bulging eyes with shallow eye sockets (ocular proptosis). These changes to the head and face become more prominent with age and are most noticeable in affected adults. In about a quarter of individuals with Camurati-Engelmann disease, the thickened skull increases pressure on the brain or compresses the spinal cord, which can cause a variety of neurological problems, including headaches, hearing loss, vision problems, dizziness (vertigo), ringing in the ears (tinnitus), and facial paralysis.
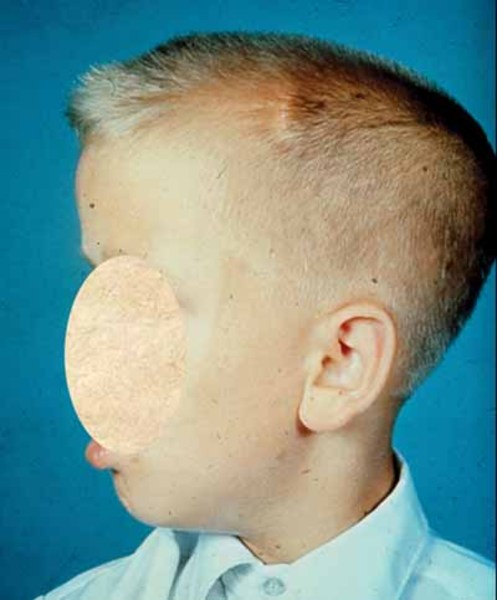
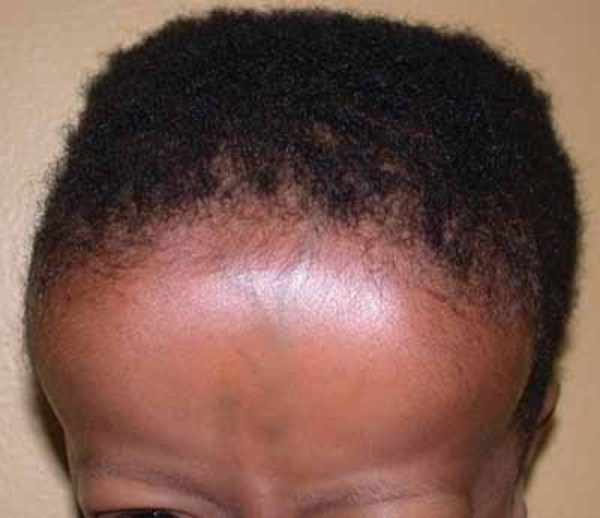
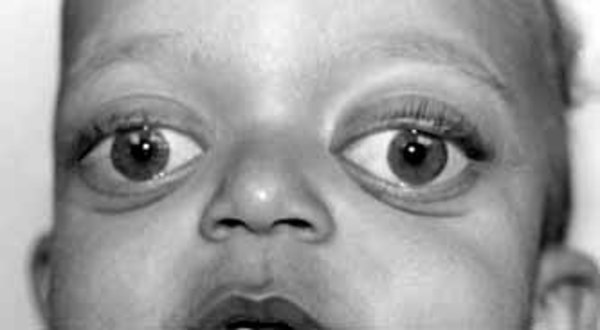
The degree of hyperostosis varies among individuals with Camurati-Engelmann disease as does the age at which they experience their first symptoms.
Other, rare features of Camurati-Engelmann disease include abnormally long limbs in proportion to height, a decrease in muscle mass and body fat, delayed teething (dentition), frequent cavities, delayed puberty, a shortage of red blood cells (anemia), an enlarged liver and spleen (hepatosplenomegaly), thinning of the skin, and excessively sweaty (hyperhidrotic) hands and feet.

Frequency
The prevalence of Camurati-Engelmann disease is unknown. More than 300 cases have been reported worldwide.
Causes
Mutations in the TGFB1 gene cause Camurati-Engelmann disease. The TGFB1 gene provides instructions for producing a protein called transforming growth factor beta-1 (TGFβ-1). The TGFβ-1 protein triggers chemical signals that regulate various cell activities, including the growth and division (proliferation) of cells, the maturation of cells to carry out specific functions (differentiation), cell movement (motility), and controlled cell death (apoptosis).
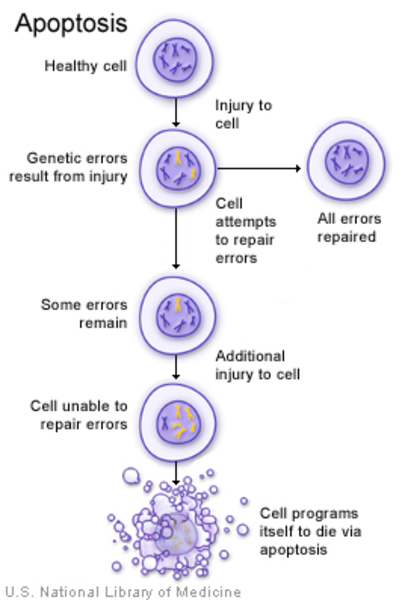
The TGFβ-1 protein is found throughout the body but is particularly abundant in tissues that make up the skeleton, where it helps regulate the formation and growth of bone and cartilage, a tough, flexible tissue that makes up much of the skeleton during early development. TGFβ-1 is involved in different processes in other tissues.
The TGFB1 gene mutations that cause Camurati-Engelmann disease result in the production of an overly active TGFβ-1 protein. This abnormal TGFβ-1 protein activity causes an increase in signaling, which leads to more bone formation. As a result, the bones in the arms, legs, and skull are thicker than normal, contributing to the movement and neurological problems often experienced by individuals with Camurati-Engelmann disease.
Some individuals with Camurati-Engelmann disease do not have an identified mutation in the TGFB1 gene. In these cases, the cause of the condition is unknown.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Some people who have the altered gene never develop the condition, a situation known as reduced penetrance.
Other Names for This Condition
- Camurati-Engelmann syndrome
- CED
- Diaphyseal dysplasia
- Diaphyseal hyperostosis
- Diaphyseal osteosclerosis
- Engelmann disease
- PDD
- Progressive diaphyseal dysplasia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Carlson ML, Beatty CW, Neff BA, Link MJ, Driscoll CL. Skull base manifestations of Camurati-Engelmann disease. Arch Otolaryngol Head Neck Surg. 2010 Jun;136(6):566-75. doi: 10.1001/archoto.2010.68. Citation on PubMed
- Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003 Feb 28;278(9):7718-24. doi: 10.1074/jbc.M208857200. Epub 2002 Dec 18. Citation on PubMed
- Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, Guanabens N, Migone N, Wientroub S, Divizia MT, Bergmann C, Bennett C, Simsek S, Melancon S, Cundy T, Van Hul W. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006 Jan;43(1):1-11. doi: 10.1136/jmg.2005.033522. Epub 2005 May 13. Citation on PubMed or Free article on PubMed Central
- Wallace SE, Lachman RS, Mekikian PB, Bui KK, Wilcox WR. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-generation pedigree, identification of a mutation in TGFB1, and review. Am J Med Genet A. 2004 Sep 1;129A(3):235-47. doi: 10.1002/ajmg.a.30148. Citation on PubMed
- Yuldashev AJ, Shin CH, Kim YS, Jang WY, Park MS, Chae JH, Yoo WJ, Choi IH, Kim OH, Cho TJ. Orthopedic Manifestations of Type I Camurati-Engelmann Disease. Clin Orthop Surg. 2017 Mar;9(1):109-115. doi: 10.4055/cios.2017.9.1.109. Epub 2017 Feb 13. Citation on PubMed or Free article on PubMed Central
- Zhao L, Hantash BM. TGF-beta1 regulates differentiation of bone marrow mesenchymal stem cells. Vitam Horm. 2011;87:127-41. doi: 10.1016/B978-0-12-386015-6.00042-1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.