

Description
Beta thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body.
In people with beta thalassemia, low levels of hemoglobin reduce oxygen levels in the body. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications. People with beta thalassemia are at an increased risk of developing abnormal blood clots.
Beta thalassemia is classified into two types depending on the severity of symptoms: thalassemia major (also known as transfusion-dependent thalassemia or Cooley's anemia) and thalassemia intermedia (which is a non-transfusion-dependent thalassemia). Of the two types, thalassemia major is more severe.
The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Puberty is delayed in some adolescents with thalassemia major.
Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems.
Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life. Affected individuals have mild to moderate anemia and may also have slow growth, bone abnormalities, and an increased risk of developing abnormal blood clots.
Frequency
Beta thalassemia is a fairly common blood disorder worldwide. Thousands of infants with beta thalassemia are born each year. Beta thalassemia occurs most frequently in people from Mediterranean countries, North Africa, the Middle East, India, Central Asia, and Southeast Asia.
Causes
Variants (also known as mutations) in the HBB gene cause beta thalassemia. The HBB gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of hemoglobin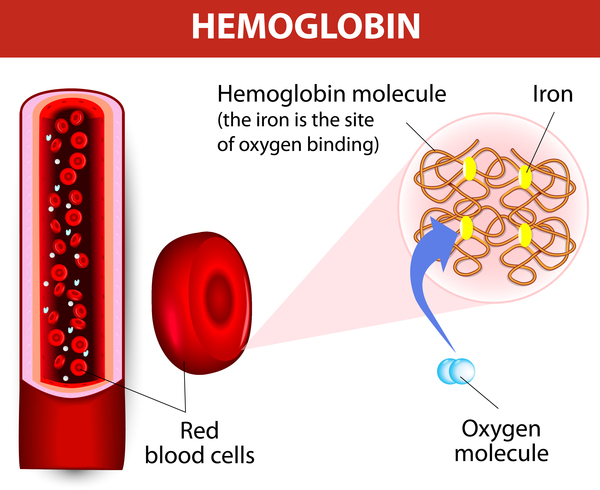 . Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin.
. Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin.
Some variants in the HBB gene prevent the production of any beta-globin. The absence of beta-globin is referred to as beta-zero (β0) thalassemia. Other HBB gene variants allow some beta-globin to be produced but in reduced amounts. A reduced amount of beta-globin is called beta-plus (β+) thalassemia. Having either β0 or β+ thalassemia does not necessarily predict disease severity, however; people with both types have been diagnosed with thalassemia major and thalassemia intermedia.
A shortage of beta-globin hinders the formation of functional hemoglobin. Without sufficient hemoglobin, red blood cells do not develop normally, causing a shortage of mature red blood cells. The low number of mature red blood cells leads to anemia and other associated health problems in people with beta thalassemia.
and other associated health problems in people with beta thalassemia.
Inheritance
Thalassemia major and thalassemia intermedia are inherited in an autosomal recessive pattern , which means both copies of the HBB gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. Sometimes, however, people with only one HBB gene variant in each cell develop mild anemia. These mildly affected people are said to have thalassemia minor.
, which means both copies of the HBB gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition. Sometimes, however, people with only one HBB gene variant in each cell develop mild anemia. These mildly affected people are said to have thalassemia minor.
In a small percentage of families, the HBB gene variant is inherited in an autosomal dominant manner . In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia.
. In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia.
Other Names for This Condition
- Erythroblastic anemia
- Mediterranean anemia
- Thalassemia, beta type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Akhavan-Niaki H, Derakhshandeh-Peykar P, Banihashemi A, Mostafazadeh A, Asghari B, Ahmadifard MR, Azizi M, Youssefi A, Elmi MM. A comprehensive molecular characterization of beta thalassemia in a highly heterogeneous population. Blood Cells Mol Dis. 2011 Jun 15;47(1):29-32. doi: 10.1016/j.bcmd.2011.03.005. Epub 2011 Apr 13. Citation on PubMed
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010 Feb;12(2):61-76. doi: 10.1097/GIM.0b013e3181cd68ed. Citation on PubMed
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 May 21;5:11. doi: 10.1186/1750-1172-5-11. Citation on PubMed or Free article on PubMed Central
- Jarjour RA, Murad H, Moasses F, Al-Achkar W. Molecular update of beta-thalassemia mutations in the Syrian population: identification of rare beta-thalassemia mutations. Hemoglobin. 2014;38(4):272-6. doi: 10.3109/03630269.2014.912661. Epub 2014 May 14. Citation on PubMed
- Langer AL. Beta-Thalassemia. 2000 Sep 28 [updated 2026 Feb 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1426/ Citation on PubMed
- National Human Genome Research Institute
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005 Sep 15;353(11):1135-46. doi: 10.1056/NEJMra050436. No abstract available. Citation on PubMed
- Thein SL. The molecular basis of beta-thalassemia. Cold Spring Harb Perspect Med. 2013 May 1;3(5):a011700. doi: 10.1101/cshperspect.a011700. Citation on PubMed or Free article on PubMed Central
- Wonke B. Clinical management of beta-thalassemia major. Semin Hematol. 2001 Oct;38(4):350-9. doi: 10.1016/s0037-1963(01)90029-0. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.