

Description
Autosomal recessive spastic ataxia of Charlevoix-Saguenay, more commonly known as ARSACS, is a condition affecting muscle movement. People with ARSACS typically have abnormal tensing of the muscles (spasticity), problems with balance and coordination (cerebellar ataxia), and reduced sensation and weakness in the arms and legs (peripheral neuropathy).
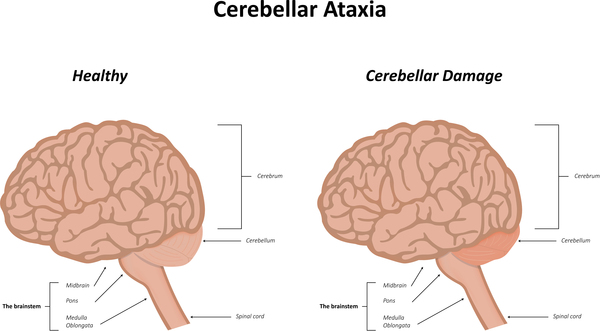
Additional muscle problems that can occur in ARSACS include muscle wasting (amyotrophy), involuntary eye movements (nystagmus), and difficulty swallowing (dysphagia) and speaking (dysarthria). Other features of ARSACS involve high-arched feet (pes cavus), a spine that curves to the side (scoliosis), yellow streaks of fatty tissue in the light-sensitive tissue at the back of the eye (hypermyelination of the retina), urinary tract problems, intellectual disability, hearing loss, and recurrent seizures (epilepsy).
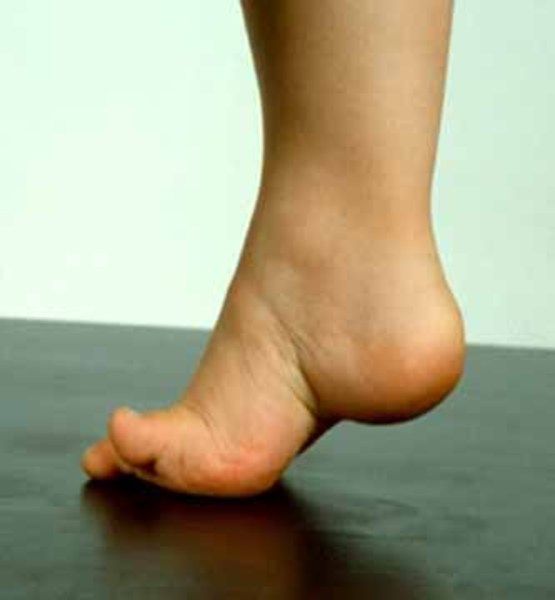


An unsteady walking style (gait) is the first symptom of ARSACS. Walking problems usually begin between the ages of 12 months and 18 months, as toddlers are learning to walk. These movement problems worsen over time, with increased spasticity and ataxia of the arms and legs. In some cases spasticity goes away, but this apparent improvement is thought to be due to the wasting away (atrophy) of nerves in the arms and legs. Most affected individuals require wheelchair assistance by the time they are in their thirties or forties.
While this condition was named after the area in which it was first seen, the Charlevoix-Saguenay region of Quebec, Canada, ARSACS has been identified in individuals worldwide.
Frequency
The incidence of ARSACS in the Charlevoix-Saguenay region is estimated to be 1 in 1,500 to 2,000 individuals. Outside of Quebec, the incidence of ARSACS is unknown. About 200 individuals with ARSACS have been described in the scientific literature.
Causes
Mutations in the SACS gene cause ARSACS. The SACS gene provides instructions for producing a protein called sacsin. Sacsin is primarily found in cells in the brain, skin, and muscles used for movement (skeletal muscles), but the specific function of the protein is unknown. Research suggests that sacsin plays a role in organizing proteins into bundles called intermediate filaments. Intermediate filaments provide support and strength to cells. In nerve cells (neurons), specialized intermediate filaments called neurofilaments comprise the structural framework that establishes the size and shape of nerve cell extensions called axons, which are essential for transmission of nerve impulses to other neurons and to muscle cells.


Mutations in the SACS gene cause the production of an unstable sacsin protein that does not function normally. It is unclear how the abnormal sacsin protein affects the brain and skeletal muscles but it likely impairs normal organization of intermediate filaments in cells, particularly neurofilaments, and disrupts neuron function. This decreased neuronal signaling may result in the signs and symptoms of ARSACS.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- ARSACS
- Charlevoix-Saguenay spastic ataxia
- Spastic ataxia of Charlevoix-Saguenay
- Spastic ataxia, Charlevoix-Saguenay type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, Bouchard JP, Mathieu J, Melancon SB, Schalling M, Lander ES, Morgan K, Hudson TJ, Richter A. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000 Feb;24(2):120-5. doi: 10.1038/72769. Citation on PubMed
- Gagnon C, Desrosiers J, Mathieu J. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: upper extremity aptitudes, functional independence and social participation. Int J Rehabil Res. 2004 Sep;27(3):253-6. doi: 10.1097/00004356-200409000-00013. Citation on PubMed
- Gagnon C, Lessard I, Lavoie C, Cote I, St-Gelais R, Mathieu J, Brais B. An exploratory natural history of ataxia of Charlevoix-Saguenay: A 2-year follow-up. Neurology. 2018 Oct 2;91(14):e1307-e1311. doi: 10.1212/WNL.0000000000006290. Epub 2018 Aug 29. Citation on PubMed or Free article on PubMed Central
- Grieco GS, Malandrini A, Comanducci G, Leuzzi V, Valoppi M, Tessa A, Palmeri S, Benedetti L, Pierallini A, Gambelli S, Federico A, Pierelli F, Bertini E, Casali C, Santorelli FM. Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology. 2004 Jan 13;62(1):103-6. doi: 10.1212/01.wnl.0000104491.66816.77. Citation on PubMed
- Ouyang Y, Segers K, Bouquiaux O, Wang FC, Janin N, Andris C, Shimazaki H, Sakoe K, Nakano I, Takiyama Y. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J Neurol Sci. 2008 Jan 15;264(1-2):73-6. doi: 10.1016/j.jns.2007.07.022. Epub 2007 Aug 22. Citation on PubMed
- Richter AM, Ozgul RK, Poisson VC, Topaloglu H. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics. 2004 Sep;5(3):165-70. doi: 10.1007/s10048-004-0179-y. Epub 2004 May 20. Citation on PubMed
- Takiyama Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropathology. 2006 Aug;26(4):368-75. doi: 10.1111/j.1440-1789.2006.00664.x. Citation on PubMed
- Takiyama Y. Sacsinopathies: sacsin-related ataxia. Cerebellum. 2007;6(4):353-9. doi: 10.1080/14734220701230466. Epub 2007 Feb 28. Citation on PubMed
- Vogel AP, Rommel N, Oettinger A, Stoll LH, Kraus EM, Gagnon C, Horger M, Krumm P, Timmann D, Storey E, Schols L, Synofzik M. Coordination and timing deficits in speech and swallowing in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). J Neurol. 2018 Sep;265(9):2060-2070. doi: 10.1007/s00415-018-8950-4. Epub 2018 Jul 2. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.