

Description
Autosomal dominant leukodystrophy with autonomic disease (ADLD) is one of a group of genetic disorders called leukodystrophies. Leukodystrophies are characterized by abnormalities of the nervous system's white matter, which consists of nerve fibers covered by a fatty substance called myelin . Myelin insulates and protects nerve fibers and promotes the rapid transmission of nerve impulses.
. Myelin insulates and protects nerve fibers and promotes the rapid transmission of nerve impulses.
People with ADLD develop signs and symptoms of the condition in adulthood, typically in their forties or fifties. The first signs of the condition often involve problems with the autonomic nervous system, which controls involuntary body processes such as the regulation of blood pressure and body temperature. These problems include difficulty with bowel and bladder function, a sharp drop in blood pressure upon standing (orthostatic hypotension), and erectile dysfunction in men. Rarely, people experience an inability to sweat (anhidrosis), which can lead to a dangerously high body temperature.
In ADLD, movement difficulties often develop after the autonomic nervous system problems. Affected individuals can have muscle stiffness (spasticity) or weakness and involuntary rhythmic shaking, called intention tremor because it worsens during movement. People with ADLD often have difficulty coordinating movements (ataxia), including movements that involve judging distance or scale (dysmetria), such as picking up a distant object, and rapidly alternating movements (dysdiadochokinesia), including hand clapping or foot stomping. These movement problems usually first affect the legs, but as the condition worsens, the arms and eventually the face become involved. In some people with ADLD, the symptoms worsen during episodes of fever, infection, or exposure to heat. Due to difficulty walking and an unsteady gait, many affected individuals need a cane, walker, or wheelchair for assistance.
Intelligence is usually unaffected; however, people who have had ADLD for a long time may have a decline in intellectual function (dementia). ADLD worsens slowly, and affected individuals usually survive 10 to 20 years after the onset of symptoms.
Frequency
The exact prevalence of ADLD is unknown. At least 70 affected individuals have been described in the scientific literature, although this condition is likely to be underdiagnosed.
Causes
ADLD is caused by mutations in the LMNB1 gene. This gene provides instructions for making the lamin B1 protein. Lamin B1 is an essential scaffolding (supporting) component of the nuclear envelope, which is the membrane that surrounds the nucleus, and plays an important role in determining the shape of the nucleus within cells. Lamin B1 also plays a role in the copying (replication) of DNA in preparation for cell division and the activity (expression) of many genes.
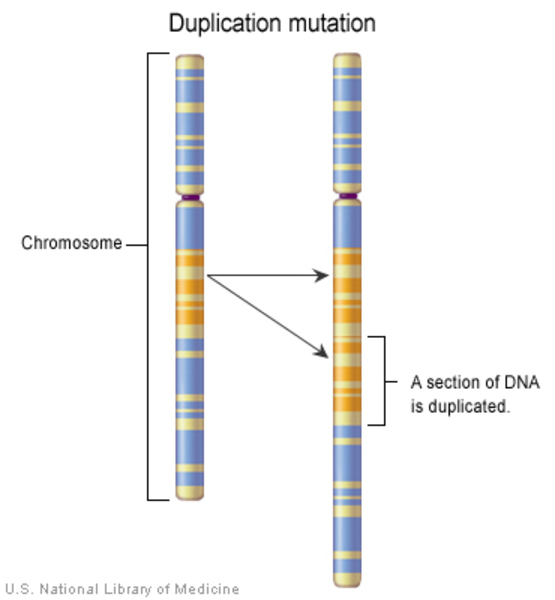
Nearly all cases of ADLD result from an abnormal extra copy (duplication) of the LMNB1 gene. As a result of this duplication, more lamin B1 is produced than normal. While lamin B1 is found in cells throughout the body, it appears that cells in the brain are especially sensitive to changes in lamin B1. Cells called oligodendrocytes, which help coat nerve cells with myelin, seem to be particularly affected. Increased lamin B1 levels lead to decreased expression of genes that play a variety of roles in the cell, including myelin production. Additionally, an increase in the amount of lamin B1 leads to a hardening of the nuclear envelope. These changes my cause problems with cell function and lead to reduced myelin production and maintenance over time.
The loss of myelin (demyelination) occurs in the brain and spinal cord (central nervous system) in people with ADLD, often years before movement problems develop. Demyelination of the spinal cord likely contributes to the early signs and symptoms of ADLD, including problems with bladder control and orthostatic hypotension, by impairing transmission of nerve signals from the brain to the body. The movement problems are probably due to demyelination in the region of the brain involved in coordinating movements (the cerebellum) and of the nerve cells that extend down the spinal cord (corticospinal tracts) and control voluntary muscle movement.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

In most cases, an affected person has one parent with the condition.
Other Names for This Condition
- ADLD
- Adult-onset autosomal dominant leukodystrophy with autonomic symptoms
- Autosomal dominant adult-onset demyelinating leukodystrophy
- LMNB1-related adult-onset autosomal dominant leukodystrophy
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bartoletti-Stella A, Gasparini L, Giacomini C, Corrado P, Terlizzi R, Giorgio E, Magini P, Seri M, Baruzzi A, Parchi P, Brusco A, Cortelli P, Capellari S. Messenger RNA processing is altered in autosomal dominant leukodystrophy. Hum Mol Genet. 2015 May 15;24(10):2746-56. doi: 10.1093/hmg/ddv034. Epub 2015 Jan 30. Citation on PubMed or Free article on PubMed Central
- Ferrera D, Canale C, Marotta R, Mazzaro N, Gritti M, Mazzanti M, Capellari S, Cortelli P, Gasparini L. Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB J. 2014 Sep;28(9):3906-18. doi: 10.1096/fj.13-247635. Epub 2014 May 22. Citation on PubMed or Free article on PubMed Central
- Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann Neurol. 2015 Sep;78(3):412-25. doi: 10.1002/ana.24452. Epub 2015 Jul 27. Citation on PubMed
- Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, Gasparini L, Ferrera D, Canale C, Guipponi M, Pennacchio LA, Antonarakis SE, Brussino A, Brusco A. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum Mol Genet. 2015 Jun 1;24(11):3143-54. doi: 10.1093/hmg/ddv065. Epub 2015 Feb 20. Citation on PubMed or Free article on PubMed Central
- Giorgio E, Rolyan H, Kropp L, Chakka AB, Yatsenko S, Di Gregorio E, Lacerenza D, Vaula G, Talarico F, Mandich P, Toro C, Pierre EE, Labauge P, Capellari S, Cortelli P, Vairo FP, Miguel D, Stubbolo D, Marques LC, Gahl W, Boespflug-Tanguy O, Melberg A, Hassin-Baer S, Cohen OS, Pjontek R, Grau A, Klopstock T, Fogel B, Meijer I, Rouleau G, Bouchard JP, Ganapathiraju M, Vanderver A, Dahl N, Hobson G, Brusco A, Brussino A, Padiath QS. Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum Mutat. 2013 Aug;34(8):1160-71. doi: 10.1002/humu.22348. Epub 2013 May 28. Citation on PubMed or Free article on PubMed Central
- Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, Fu YH. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013 Jun;123(6):2719-29. doi: 10.1172/JCI66737. Epub 2013 May 15. Citation on PubMed or Free article on PubMed Central
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006 Oct;38(10):1114-23. doi: 10.1038/ng1872. Epub 2006 Sep 3. Citation on PubMed
- Rolyan H, Tyurina YY, Hernandez M, Amoscato AA, Sparvero LJ, Nmezi BC, Lu Y, Estecio MR, Lin K, Chen J, He RR, Gong P, Rigatti LH, Dupree J, Bayir H, Kagan VE, Casaccia P, Padiath QS. Defects of Lipid Synthesis Are Linked to the Age-Dependent Demyelination Caused by Lamin B1 Overexpression. J Neurosci. 2015 Aug 26;35(34):12002-17. doi: 10.1523/JNEUROSCI.1668-15.2015. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.