

Description
Androgen insensitivity syndrome is a condition that affects sexual development before birth and during puberty. People with this condition have one X chromosome and one Y chromosome in each cell. In people with androgen insensitivity syndrome, the body's cells and tissues are unable to respond to certain male sex hormones (called androgens) that are important for normal male sexual development before birth and during puberty. As a result, affected individuals may have external sex characteristics that are typical for females or have features of both male and female sexual development.
in each cell. In people with androgen insensitivity syndrome, the body's cells and tissues are unable to respond to certain male sex hormones (called androgens) that are important for normal male sexual development before birth and during puberty. As a result, affected individuals may have external sex characteristics that are typical for females or have features of both male and female sexual development.
There are three forms of androgen insensitivity syndrome: complete, partial, and mild.
Complete androgen insensitivity syndrome occurs when the body does not respond to androgens at all. People with this form of the condition have external sex characteristics that are typical of females. Affected individuals do not have a uterus. They have male internal sex organs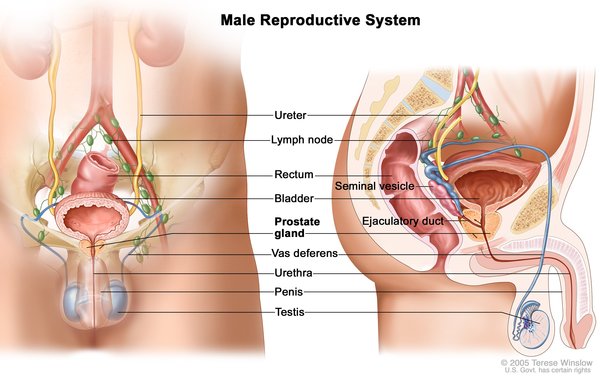 (testes) that are undescended, which means they are located in the pelvis or abdomen instead of outside the body. As such, affected individuals do not menstruate and are unable to conceive a child (infertile). People with complete androgen insensitivity syndrome also have sparse or absent hair in the pubic area and under the arms.
(testes) that are undescended, which means they are located in the pelvis or abdomen instead of outside the body. As such, affected individuals do not menstruate and are unable to conceive a child (infertile). People with complete androgen insensitivity syndrome also have sparse or absent hair in the pubic area and under the arms.
The partial and mild forms of androgen insensitivity syndrome occur when the body's tissues are partially sensitive to the effects of androgens.
People with partial androgen insensitivity can have genitalia that look typical for females, genitalia that have both male and female characteristics, or genitalia that look typical for males.
People with mild androgen insensitivity are born with male-typical sex characteristics, but they are often infertile and tend to experience breast enlargement at puberty.
Frequency
Complete androgen insensitivity syndrome affects 2 to 5 in 100,000 newborn females. Partial androgen insensitivity is thought to be at least as common as complete androgen insensitivity. Mild androgen insensitivity is much less common.
Causes
Variants (also called mutations) in the AR gene cause androgen insensitivity syndrome. This gene provides instructions for making a protein called an androgen receptor. Androgen receptor proteins interact with androgen hormones, such as testosterone, and help direct male sexual development. Specifically, the androgen receptor attaches (binds) to androgen hormones to form an androgen-receptor complex. This complex then binds to DNA to regulate the activity of certain genes that play a role in male sexual development. Androgens and androgen receptors also have other important functions in both males and females, such as regulating hair growth and sex drive.
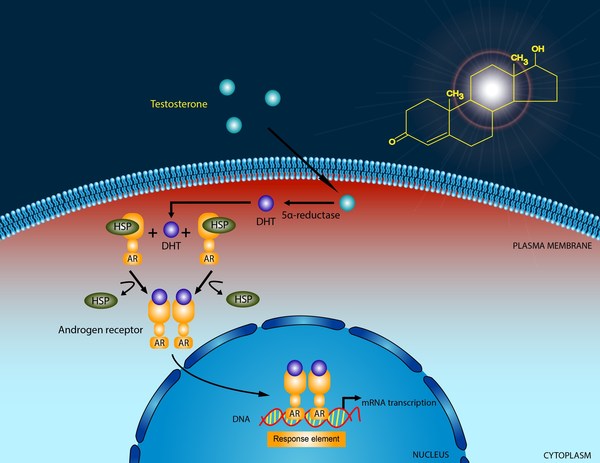
Variants in the AR gene prevent androgen receptors from working properly, which makes them less able to bind to testosterone and regulate gene activity. If androgen receptors cannot bind to androgens, the body cannot use androgens, even if there are normal levels of these hormones in the body.
Inheritance
This condition is inherited in an X-linked recessive pattern . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that both copies of a gene would be altered, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that both copies of a gene would be altered, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
About 70 percent of all cases of androgen insensitivity syndrome are inherited from people who carry an altered copy of the AR gene on one of their two X chromosomes. The remaining 30 percent of cases result from new (de novo) variants in the gene that occur in the egg cell before the child is conceived or during early embryonic development.
Other Names for This Condition
- AIS
- Androgen receptor deficiency
- Androgen resistance syndrome
- AR deficiency
- DHTR deficiency
- Dihydrotestosterone receptor deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chen MJ, Vu BM, Axelrad M, Dietrich JE, Gargollo P, Gunn S, Macias CG, McCullough LB, Roth DR, Sutton VR, Karaviti LP. Androgen Insensitivity Syndrome: Management Considerations from Infancy to Adulthood. Pediatr Endocrinol Rev. 2015 Jun;12(4):373-87. Citation on PubMed
- Delli Paoli E, Di Chiano S, Paoli D, Lenzi A, Lombardo F, Pallotti F. Androgen insensitivity syndrome: a review. J Endocrinol Invest. 2023 Nov;46(11):2237-2245. doi: 10.1007/s40618-023-02127-y. Epub 2023 Jun 10. Citation on PubMed
- Gottlieb B, Pinsky L, Beitel LK, Trifiro M. Androgen insensitivity. Am J Med Genet. 1999 Dec 29;89(4):210-7. doi: 10.1002/(sici)1096-8628(19991229)89:43.0.co;2-p. Citation on PubMed
- Gottlieb B, Trifiro MA. Androgen Insensitivity Syndrome. 1999 Mar 24 [updated 2017 May 11]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1429/ Citation on PubMed
- Hiort O. Clinical and molecular aspects of androgen insensitivity. Endocr Dev. 2013;24:33-40. doi: 10.1159/000342499. Epub 2013 Feb 1. Citation on PubMed
- Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet. 2012 Oct 20;380(9851):1419-28. doi: 10.1016/S0140-6736(12)60071-3. Epub 2012 Jun 13. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.