

Description
Alkaptonuria is an inherited condition that causes arthritis, kidney stones, spots of dark pigmentation, and dark urine. Ochronosis, a buildup of dark (blue-black) pigment in certain tissues, is a characteristic feature of alkaptonuria. The first symptom of alkaptonuria is often urine that turns black or very dark when it is exposed to air (oxidation). However, this color change may not occur immediately after urination.
Ochronosis occurs in connective tissues throughout the body, such as the joints, tendons, and ligaments. Pigment changes can be easily seen in the whites of the eyes (sclera), the outer ears, and the hands. Dark pigment can also be found in earwax and in body sweat (perspiration). These pigment changes are usually evident after age 30.
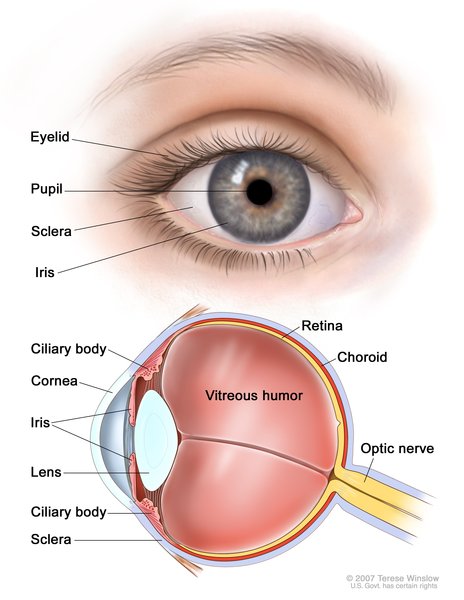

Ochronosis in the joints causes people with alkaptonuria to develop arthritis, typically in early adulthood. Arthritis usually affects the spine and large joints (such as the hips and knees). Many affected individuals require joint replacements later in life.
Over time, the deposits of pigment can harden (calcify). Calcification in joints or ligaments causes them to become rigid and brittle, decreasing their flexibility and making them prone to damage. Calcification of the discs that separate the bones of the spine can cause further back pain. This calcification can also affect the cartilage that makes up the valves of the heart, which allow blood to move through the heart and prevent blood from flowing backward. As a result, the heart valves narrow (stenosis), which can cause the valves to leak (regurgitation).
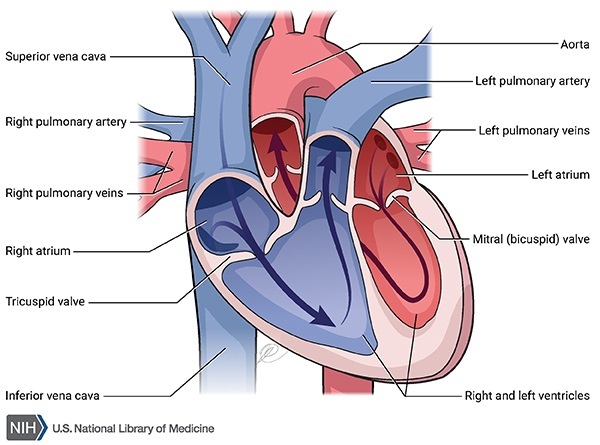
The body removes the excess pigment in urine. However, removing the large pigment deposits found in people with alkaptonuria can put strain on the kidneys, and this can lead to kidney (renal) failure. Similarly, a buildup of these substances in the kidneys or prostate gland can cause calcium stones to develop. If the body cannot effectively process and remove the pigment and related substances, these products substances can continue to build up in the body, worsening ochronosis, arthritis, and other features of alkaptonuria.

With appropriate medical management, people with alkaptonuria generally have a normal life expectancy.
Frequency
Alkaptonuria is rare, affecting 1 in 250,000 to 1 million people in the United States. Alkaptonuria is more common in certain areas of Slovakia (where it has an incidence of about 1 in 19,000 people) and in the Dominican Republic.
Causes
Variants (also called mutations) in the HGD gene cause alkaptonuria. The HGD gene provides instructions for making an enzyme called homogentisate 1,2-dioxygenase. This enzyme helps break down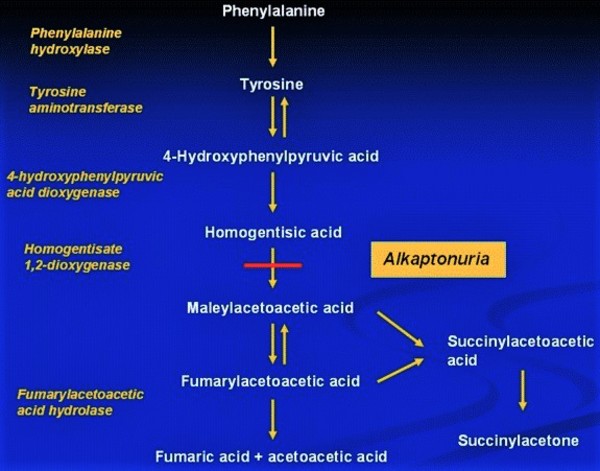 the amino acids phenylalanine and tyrosine, which are important building blocks of proteins. Phenylalanine and tyrosine are broken down when they are no longer needed or when there are too many of them.
the amino acids phenylalanine and tyrosine, which are important building blocks of proteins. Phenylalanine and tyrosine are broken down when they are no longer needed or when there are too many of them.
Variants in the HGD gene cause the cell to produce a version of the enzyme that does not function well. As a result, phenylalanine and tyrosine are not completely broken down. Instead, a substance called homogentisic acid, which is produced during the breakdown process, accumulates in the body.
Excess homogentisic acid settles in connective tissues, forming a substance called ochronotic pigment, which causes cartilage and skin to darken. Over time, a buildup of this pigment calcifies, leading to many of the health problems seen in people with alkaptonuria. Excess homogentisic acid is also excreted in urine, making urine turn dark when exposed to air.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- AKU
- Alcaptonuria
- Homogentisic acid oxidase deficiency
- Homogentisic acidura
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bernardini G, Braconi D, Zatkova A, Sireau N, Kujawa MJ, Introne WJ, Spiga O, Geminiani M, Gallagher JA, Ranganath LR, Santucci A. Alkaptonuria. Nat Rev Dis Primers. 2024 Mar 7;10(1):16. doi: 10.1038/s41572-024-00498-x. Citation on PubMed
- Bernini A, Spiga O, Santucci A. Structure-Function Relationship of Homogentisate 1,2-dioxygenase: Understanding the Genotype-Phenotype Correlations in the Rare Genetic Disease Alkaptonuria. Curr Protein Pept Sci. 2023;24(5):380-392. doi: 10.2174/1389203724666230307104135. Citation on PubMed
- Davison AS, Norman BP. Alkaptonuria - Past, present and future. Adv Clin Chem. 2023;114:47-81. doi: 10.1016/bs.acc.2023.02.005. Epub 2023 Mar 28. Citation on PubMed
- Introne WJ, Perry M, Chen M. Alkaptonuria. 2003 May 9 [updated 2021 Jun 10]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1454/ Citation on PubMed
- Milella MS, Geminiani M, Trezza A, Visibelli A, Braconi D, Santucci A. Alkaptonuria: From Molecular Insights to a Dedicated Digital Platform. Cells. 2024 Jun 20;13(12):1072. doi: 10.3390/cells13121072. Citation on PubMed
- Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, Gahl WA. Natural history of alkaptonuria. N Engl J Med. 2002 Dec 26;347(26):2111-21. doi: 10.1056/NEJMoa021736. Citation on PubMed
- Zatkova A. An update on molecular genetics of Alkaptonuria (AKU). J Inherit Metab Dis. 2011 Dec;34(6):1127-36. doi: 10.1007/s10545-011-9363-z. Epub 2011 Jul 1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.