

Description
Aicardi-Goutières syndrome is a disorder with variable signs and symptoms, but it primarily affects the brain, the immune system, and the skin.
Aicardi-Goutières syndrome is often divided into two types, which are distinguished by the severity of features and the age at which they begin: the early-onset form (sometimes called the classic form) and the later-onset form.
Individuals with the early-onset form of Aicardi-Goutières syndrome can experience severe brain dysfunction (encephalopathy) within the first months of life. This encephalopathic phase of the disorder can last for weeks or months. Affected infants stop developing new skills and begin losing skills they had already acquired (developmental regression). Infants with this form can have seizures. Medical imaging reveals loss of white matter in the brain (leukodystrophy). White matter consists of nerve cells covered by myelin, which is a substance that protects nerves and allows them to rapidly transmit nerve impulses. Growth of the brain and skull slows down, resulting in an abnormally small head size (microcephaly). Affected individuals may have abnormal deposits of calcium (calcification) in the brain. As a result of this neurological damage, most people with Aicardi-Goutières syndrome have profound intellectual disabilities.

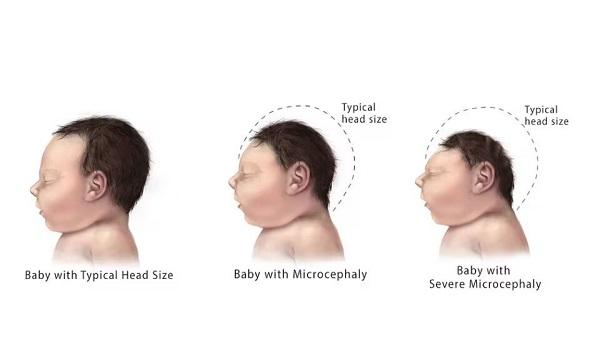
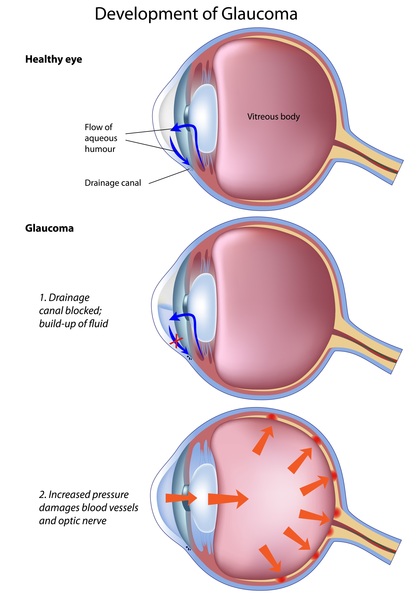
Affected babies are usually extremely irritable and do not feed well. They also have muscle stiffness (spasticity), involuntary tensing of various muscles (dystonia), and weak muscle tone (hypotonia). They can have vision problems including vision loss and increased pressure in the eye (glaucoma).
Some newborns have a combination of features that include an enlarged liver and spleen (hepatosplenomegaly), elevated blood levels of liver enzymes, and a shortage of blood cells called platelets that are needed for normal blood clotting (thrombocytopenia). They may develop intermittent fevers in the absence of infection (sterile pyrexias). While this combination of signs and symptoms is typically associated with the immune system's response to a viral infection that is present at birth (congenital), no actual infection is found in these infants. For this reason, Aicardi-Goutières syndrome is sometimes referred to as a "mimic of congenital infection."
In some affected newborns, white blood cells, interferon proteins, and other immune system molecules can be detected in the cerebrospinal fluid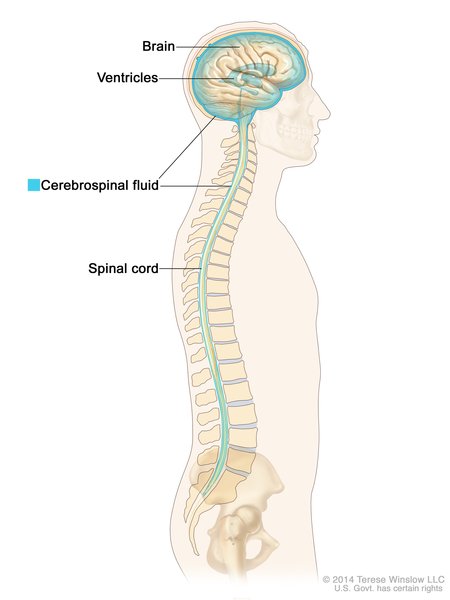 , which is the fluid that surrounds the brain and spinal cord (central nervous system). These findings are consistent with inflammation and tissue damage
, which is the fluid that surrounds the brain and spinal cord (central nervous system). These findings are consistent with inflammation and tissue damage in the central nervous system.
in the central nervous system.
About 40 percent of people with the early-onset form of Aicardi-Goutières syndrome develop a skin problem called chilblains. Chilblains are painful, itchy skin lesions that are puffy and red, and they usually appear on the fingers, toes, nose, and ears. They are caused by inflammation of small blood vessels and may be brought on or made worse by exposure to cold temperatures.
In about 20 percent of cases, the early-onset form of Aicardi-Goutières syndrome begins prenatally. Slow growth (intrauterine growth retardation) and brain abnormalities, especially brain calcification, may be seen on ultrasound imaging. These individuals have the most severe neurological problems and the highest risk for early death.
People with the later-onset form of Aicardi-Goutières syndrome typically have normal development in infancy. In these individuals, encephalopathy typically occurs after 1 year of age. Similar to those with the early-onset form, babies with the later-onset form experience irritability, poor feeding, and sterile pyrexias. Over time, affected individuals show developmental delays and regression. They may also have spasticity and hypotonia, and the growth of the brain and head may slow leading to microcephaly. The health and developmental problems in people with the later-onset form are typically not as severe as those in individuals with the early-onset form, though the severity can vary among affected individuals.
As a result of the severe neurological problems that are usually associated with Aicardi-Goutières syndrome, most people with this disorder do not survive past childhood. However, some affected individuals with the later-onset form of the condition and milder neurological problems can live into adolescence or adulthood.
Frequency
Aicardi-Goutières syndrome is a rare disorder. More than 500 people with Aicardi-Goutières syndrome have been described in the scientific literature, though the exact prevalence of the condition is unknown.
Causes
Variants (also called mutations) in several genes can cause Aicardi-Goutières syndrome. The TREX1, RNASEH2A, RNASEH2B, and RNASEH2C genes, provide instructions for making nucleases, which are enzymes that help break down molecules of DNA and its chemical cousin RNA when they are no longer needed. These DNA and RNA molecules or fragments may be generated during the first stage of protein production (transcription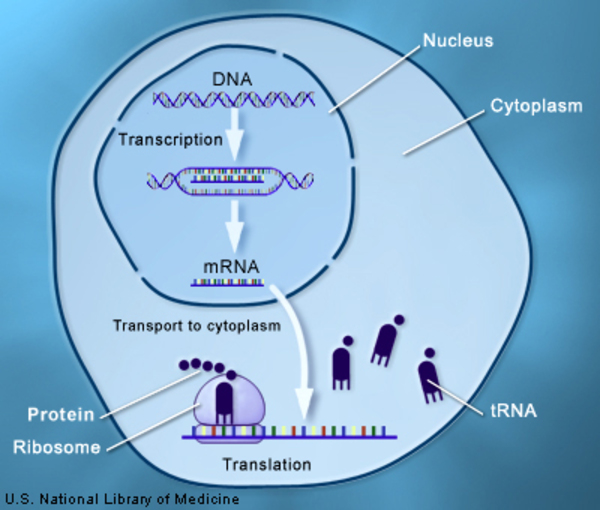 ), the replication of cells' genetic material that occurs when the cell is preparing for cell division, DNA repair, cell death (apoptosis), and other processes. The nuclease enzymes produced by genes with variants may not function properly, and some variants may prevent the enzyme from being produced at all. Researchers suggest that this may result in the accumulation of unneeded DNA and RNA in cells. The unneeded DNA and RNA may be mistaken by cells for the genetic material of viral invaders. This triggers an immune system reaction that includes the abnormal activation of interferon proteins, which play a critical role in the immune system including regulating inflammation. The abnormal immune response results in encephalopathy, skin lesions, and other signs and symptoms of Aicardi-Goutières syndrome.
), the replication of cells' genetic material that occurs when the cell is preparing for cell division, DNA repair, cell death (apoptosis), and other processes. The nuclease enzymes produced by genes with variants may not function properly, and some variants may prevent the enzyme from being produced at all. Researchers suggest that this may result in the accumulation of unneeded DNA and RNA in cells. The unneeded DNA and RNA may be mistaken by cells for the genetic material of viral invaders. This triggers an immune system reaction that includes the abnormal activation of interferon proteins, which play a critical role in the immune system including regulating inflammation. The abnormal immune response results in encephalopathy, skin lesions, and other signs and symptoms of Aicardi-Goutières syndrome.
Variants in other genes, including the SAMHD1, IFIH1, and ADAR genes, can also cause Aicardi-Goutières syndrome. These genes provide instructions for making proteins that are involved in the immune system. Variants in these genes cause inappropriate activation of interferon proteins and the body's immune system, resulting in inflammatory damage to the brain, skin, and other body systems that leads to the characteristic features of Aicardi-Goutières syndrome.
Variants in other genes, including the RNU7-1 and LSM11 genes, can also cause Aicardi-Goutières syndrome. These genes provide instructions for making proteins that play a critical role in the cell cycle, DNA replication, and protein production. Variants in these genes disrupt these processes, causing DNA to build up in cells. This excessive amount of DNA may be mistaken for viral invaders, triggering activation of interferon proteins and other immune system reactions that lead to the other signs and symptoms of Aicardi-Goutières syndrome.
Variants in the TREX1, RNASEH2A, and RNASEH2C genes tend to cause the early-onset form of Aicardi-Goutières syndrome, while variants in the RNASEH2B, SAMHD1, IFIH1, and ADAR genes tend to cause the later-onset form. However, not every case of Aicardi-Goutières syndrome follows this pattern.
Because the signs and symptoms of Aicardi-Goutières syndrome are caused in part by abnormal activation of interferon proteins, it one of a group of disorders known as interferonopathies.
Inheritance
Aicardi-Goutières syndrome can have different inheritance patterns. In most cases, it is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. Autosomal recessive inheritance occurs when Aicardi-Goutières syndrome is caused by variants in the ADAR, LSM11, RNASEH2A, RNASEH2B, RNASEH2C, RNU7-1, SAMHD1, and TREX1 genes. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. Autosomal recessive inheritance occurs when Aicardi-Goutières syndrome is caused by variants in the ADAR, LSM11, RNASEH2A, RNASEH2B, RNASEH2C, RNU7-1, SAMHD1, and TREX1 genes. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
When Aicardi-Goutières syndrome is caused by variants in the IFIH1 gene or by certain variants in the TREX1 or ADAR gene, it is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Other Names for This Condition
- AGS
- Aicardi Goutieres syndrome
- Cree encephalitis
- Encephalopathy with basal ganglia calcification
- Familial infantile encephalopathy with intracranial calcification and chronic cerebrospinal fluid lymphocytosis
- Pseudotoxoplasmosis syndrome
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Aicardi Goutieres syndrome

- Genetic Testing Registry: Aicardi-Goutieres syndrome 6
- Genetic Testing Registry: Aicardi-Goutieres syndrome 7
- Genetic Testing Registry: Aicardi-Goutieres syndrome 1
- Genetic Testing Registry: Aicardi-Goutieres syndrome 2
- Genetic Testing Registry: Aicardi-Goutieres syndrome 3
- Genetic Testing Registry: Aicardi-Goutieres syndrome 4
- Genetic Testing Registry: Aicardi-Goutieres syndrome 5
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- AICARDI-GOUTIERES SYNDROME 1; AGS1
- AICARDI-GOUTIERES SYNDROME 2; AGS2
- AICARDI-GOUTIERES SYNDROME 3; AGS3
- AICARDI-GOUTIERES SYNDROME 4; AGS4
- AICARDI-GOUTIERES SYNDROME 7; AGS7
- AICARDI-GOUTIERES SYNDROME 5; AGS5
- AICARDI-GOUTIERES SYNDROME 6; AGS6
- AICARDI-GOUTIERES SYNDROME 8; AGS8
- AICARDI-GOUTIERES SYNDROME 9; AGS9
Scientific Articles on PubMed
References
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-Shalev S, Arellano M, Ariaudo G, Aswani V, Babul-Hirji R, Baildam EM, Bahi-Buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Beresford MW, Bernard G, Bianchi M, Billette de Villemeur T, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-Gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Sierra Corcoles C, Cordeiro NJ, Crichiutti G, Dabydeen L, Dale RC, D'Arrigo S, De Goede CG, De Laet C, De Waele LM, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Fazzi E, Ferrie CD, Figueiredo A, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Kara B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Landrieu P, Lauffer H, Laugel V, La Piana R, Lim MJ, Lin JP, Linnankivi T, Mackay MT, Marom DR, Marques Lourenco C, McKee SA, Moroni I, Morton JE, Moutard ML, Murray K, Nabbout R, Nampoothiri S, Nunez-Enamorado N, Oades PJ, Olivieri I, Ostergaard JR, Perez-Duenas B, Prendiville JS, Ramesh V, Rasmussen M, Regal L, Ricci F, Rio M, Rodriguez D, Roubertie A, Salvatici E, Segers KA, Sinha GP, Soler D, Spiegel R, Stodberg TI, Straussberg R, Swoboda KJ, Suri M, Tacke U, Tan TY, te Water Naude J, Wee Teik K, Thomas MM, Till M, Tonduti D, Valente EM, Van Coster RN, van der Knaap MS, Vassallo G, Vijzelaar R, Vogt J, Wallace GB, Wassmer E, Webb HJ, Whitehouse WP, Whitney RN, Zaki MS, Zuberi SM, Livingston JH, Rozenberg F, Lebon P, Vanderver A, Orcesi S, Rice GI. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015 Feb;167A(2):296-312. doi: 10.1002/ajmg.a.36887. Epub 2015 Jan 16. Citation on PubMed or Free article on PubMed Central
- Crow YJ. Aicardi-Goutieres Syndrome. 2005 Jun 29 [updated 2016 Nov 22]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1475/ Citation on PubMed
- Crow YJ. Aicardi-Goutieres syndrome. Handb Clin Neurol. 2013;113:1629-35. doi: 10.1016/B978-0-444-59565-2.00031-9. Citation on PubMed
- Fazzi E, Cattalini M, Orcesi S, Tincani A, Andreoli L, Balottin U, De Simone M, Fredi M, Facchetti F, Galli J, Giliani S, Izzotti A, Meini A, Olivieri I, Plebani A. Aicardi-Goutieres syndrome, a rare neurological disease in children: a new autoimmune disorder? Autoimmun Rev. 2013 Feb;12(4):506-9. doi: 10.1016/j.autrev.2012.08.012. Epub 2012 Aug 24. Citation on PubMed
- Goutieres F. Aicardi-Goutieres syndrome. Brain Dev. 2005 Apr;27(3):201-6. doi: 10.1016/j.braindev.2003.12.011. Citation on PubMed
- Livingston JH, Crow YJ. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics. 2016 Dec;47(6):355-360. doi: 10.1055/s-0036-1592307. Epub 2016 Sep 19. Citation on PubMed
- Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, Richardt HH, Buckard J, Bevot A, Siegel C, von Stulpnagel C, Ikonomidou C, Thomas K, Proud V, Niemann F, Wieczorek D, Hausler M, Niggemann P, Baltaci V, Conrad K, Lebon P, Lee-Kirsch MA. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010 May;62(5):1469-77. doi: 10.1002/art.27367. Citation on PubMed
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D'Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007 Oct;81(4):713-25. doi: 10.1086/521373. Epub 2007 Sep 4. Citation on PubMed or Free article on PubMed Central
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009 Jul;41(7):829-32. doi: 10.1038/ng.373. Epub 2009 Jun 14. Citation on PubMed or Free article on PubMed Central
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, Del Toro M, Effat L, Enamorado NN, Fazzi E, Gener B, Haldre M, Lin JP, Livingston JH, Lourenco CM, Marques W Jr, Oades P, Peterson P, Rasmussen M, Roubertie A, Schmidt JL, Shalev SA, Simon R, Spiegel R, Swoboda KJ, Temtamy SA, Vassallo G, Vilain CN, Vogt J, Wermenbol V, Whitehouse WP, Soler D, Olivieri I, Orcesi S, Aglan MS, Zaki MS, Abdel-Salam GM, Vanderver A, Kisand K, Rozenberg F, Lebon P, Crow YJ. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013 Dec;12(12):1159-69. doi: 10.1016/S1474-4422(13)70258-8. Epub 2013 Oct 30. Citation on PubMed or Free article on PubMed Central
- Stephenson JB. Aicardi-Goutieres syndrome (AGS). Eur J Paediatr Neurol. 2008 Sep;12(5):355-8. doi: 10.1016/j.ejpn.2007.11.010. Epub 2008 Mar 14. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.