

Description
46,XX testicular difference of sex development is a condition in which individuals with two X chromosomes in each cell, the pattern typically found in females, have a male appearance. People with this condition have male external genitalia . They generally have small testes and may also have other features such as undescended testes (cryptorchidism) or the urethra opening on the underside of the penis (hypospadias). A small number of affected people have external genitalia that do not look clearly male or clearly female.
. They generally have small testes and may also have other features such as undescended testes (cryptorchidism) or the urethra opening on the underside of the penis (hypospadias). A small number of affected people have external genitalia that do not look clearly male or clearly female.
At puberty, most affected individuals require treatment with the male sex hormone testosterone to induce development of male secondary sex characteristics such as facial hair and deepening of the voice (masculinization). Hormone treatment can also help prevent breast enlargement (gynecomastia). Adults with this condition are usually shorter than average for males and are unable to have children (infertile).
Frequency
Approximately 1 in 20,000 individuals with a male appearance have 46,XX testicular difference of sex development.
Causes
People normally have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male-typical or female-typical sex characteristics. Females usually have two X chromosomes (46,XX), and males usually have one X chromosome and one Y chromosome (46,XY).
because they help determine whether a person will develop male-typical or female-typical sex characteristics. Females usually have two X chromosomes (46,XX), and males usually have one X chromosome and one Y chromosome (46,XY).
The SRY gene, normally located on the Y chromosome, provides instructions for making the sex-determining region Y protein. The sex-determining region Y protein causes a fetus to develop as a male.
In about 80 percent of individuals with 46,XX testicular difference of sex development, the condition results from an abnormal exchange of genetic material between chromosomes (translocation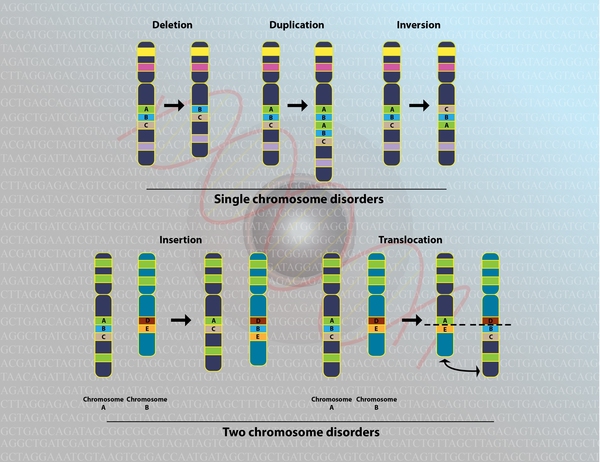 ). This exchange occurs as a random event during the formation of sperm cells
). This exchange occurs as a random event during the formation of sperm cells in the affected person's father. The translocation causes the SRY gene to be misplaced, almost always onto an X chromosome. If a fetus is conceived from a sperm cell with an X chromosome bearing the SRY gene, it will develop as a male despite not having a Y chromosome. This form of the condition is called SRY-positive 46,XX testicular difference of sex development.
in the affected person's father. The translocation causes the SRY gene to be misplaced, almost always onto an X chromosome. If a fetus is conceived from a sperm cell with an X chromosome bearing the SRY gene, it will develop as a male despite not having a Y chromosome. This form of the condition is called SRY-positive 46,XX testicular difference of sex development.
About 20 percent of people with 46,XX testicular difference of sex development do not have the SRY gene. This form of the condition is called SRY-negative 46,XX testicular difference of sex development. The cause of the condition in these individuals is often unknown, although changes affecting other genes involved in the development of sex characteristics have been identified in a small number of people with the condition. Individuals with SRY-negative 46,XX testicular difference of sex development are more likely to have genitalia that do not clearly look male or female than are people with the SRY-positive form.
Inheritance
SRY-positive 46,XX testicular difference of sex development is almost never inherited. This condition results from the translocation of a Y chromosome segment containing the SRY gene during the formation of sperm (spermatogenesis). Affected people typically have no history of the condition in their family and cannot pass on the condition because they are infertile.
In rare cases, the SRY gene may be misplaced onto a chromosome other than the X chromosome. This translocation may be carried by an unaffected father and passed on to a child with two X chromosomes, resulting in 46,XX testicular difference of sex development. In another very rare situation, a male may carry the SRY gene on both the X and Y chromosome; a child who inherits his X chromosome will develop male sex characteristics despite having no Y chromosome.
The inheritance pattern of SRY-negative 46,XX testicular difference of sex development is variable. In affected people with a known genetic cause, the condition often follows an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. In some cases, the genetic variant (also known as a mutation) is inherited from an unaffected parent. Usually, the variant is inherited from an unaffected father. Sometimes, the variant is inherited from an unaffected mother. When some people with the variant do not develop features of the condition, it is said to have reduced (or incomplete) penetrance.
pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. In some cases, the genetic variant (also known as a mutation) is inherited from an unaffected parent. Usually, the variant is inherited from an unaffected father. Sometimes, the variant is inherited from an unaffected mother. When some people with the variant do not develop features of the condition, it is said to have reduced (or incomplete) penetrance.
Other cases of SRY-negative 46,XX testicular difference of sex development result from new (de novo) variants in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the condition in their family.
in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the condition in their family.
The inheritance pattern of SRY-negative 46,XX testicular difference of sex development without an identified genetic cause is unknown. A few families with unaffected parents have had more than one child with the condition, suggesting the possibility of autosomal recessive inheritance. Autosomal recessive means both copies of a gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
inheritance. Autosomal recessive means both copies of a gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- 46,XX testicular disorder of sex development
- 46,XX testicular DSD
- nonsyndromic 46,XX testicular disorder/difference of sex development
- XX male syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abbas N, McElreavey K, Leconiat M, Vilain E, Jaubert F, Berger R, Nihoul-Fekete C, Rappaport R, Fellous M. Familial case of 46,XX male and 46,XX true hermaphrodite associated with a paternal-derived SRY-bearing X chromosome. C R Acad Sci III. 1993;316(4):375-83. Citation on PubMed
- Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, Buonocore F, Barseghyan H, Bingham N, Rosenfeld JA, Mulukutla SN, Jain M, Burrage L, Dhar S, Balasubramanyam A, Lee B; Members of UDN; Dumargne MC, Eozenou C, Suntharalingham JP, de Silva K, Lin L, Bignon-Topalovic J, Poulat F, Lagos CF, McElreavey K, Achermann JC. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. 2016 Aug 15;25(16):3446-3453. doi: 10.1093/hmg/ddw186. Epub 2016 Jul 4. Citation on PubMed
- Delot EC, Vilain EJ. Nonsyndromic 46,XX Testicular Disorders/Differences of Sex Development. 2003 Oct 30 [updated 2022 May 26]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1416/ Citation on PubMed
- Ergun-Longmire B, Vinci G, Alonso L, Matthew S, Tansil S, Lin-Su K, McElreavey K, New MI. Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab. 2005 Aug;18(8):739-48. doi: 10.1515/jpem.2005.18.8.739. Citation on PubMed
- Grigorescu-Sido A, Heinrich U, Grigorescu-Sido P, Jauch A, Hager HD, Vogt PH, Duncea I, Bettendorf M. Three new 46,XX male patients: a clinical, cytogenetic and molecular analysis. J Pediatr Endocrinol Metab. 2005 Feb;18(2):197-203. doi: 10.1515/jpem.2005.18.2.197. Citation on PubMed
- Kim GJ, Sock E, Buchberger A, Just W, Denzer F, Hoepffner W, German J, Cole T, Mann J, Seguin JH, Zipf W, Costigan C, Schmiady H, Rostasy M, Kramer M, Kaltenbach S, Rosler B, Georg I, Troppmann E, Teichmann AC, Salfelder A, Widholz SA, Wieacker P, Hiort O, Camerino G, Radi O, Wegner M, Arnold HH, Scherer G. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J Med Genet. 2015 Apr;52(4):240-7. doi: 10.1136/jmedgenet-2014-102864. Epub 2015 Jan 20. Citation on PubMed
- Kolon TF, Ferrer FA, McKenna PH. Clinical and molecular analysis of XX sex reversed patients. J Urol. 1998 Sep;160(3 Pt 2):1169-72; discussion 1178. doi: 10.1097/00005392-199809020-00057. Citation on PubMed
- Kremen J, Chan YM, Swartz JM. Recent findings on the genetics of disorders of sex development. Curr Opin Urol. 2017 Jan;27(1):1-6. doi: 10.1097/MOU.0000000000000353. Citation on PubMed
- Queralt R, Madrigal I, Vallecillos MA, Morales C, Ballesca JL, Oliva R, Soler A, Sanchez A, Margarit E. Atypical XX male with the SRY gene located at the long arm of chromosome 1 and a 1qter microdeletion. Am J Med Genet A. 2008 May 15;146A(10):1335-40. doi: 10.1002/ajmg.a.32284. Citation on PubMed
- Rajender S, Rajani V, Gupta NJ, Chakravarty B, Singh L, Thangaraj K. SRY-negative 46,XX male with normal genitals, complete masculinization and infertility. Mol Hum Reprod. 2006 May;12(5):341-6. doi: 10.1093/molehr/gal030. Epub 2006 Mar 23. Citation on PubMed
- Rizvi AA. 46, XX man with SRY gene translocation: cytogenetic characteristics, clinical features and management. Am J Med Sci. 2008 Apr;335(4):307-9. doi: 10.1097/MAJ.0b013e31811ec1b4. Citation on PubMed
- Vorona E, Zitzmann M, Gromoll J, Schuring AN, Nieschlag E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J Clin Endocrinol Metab. 2007 Sep;92(9):3458-65. doi: 10.1210/jc.2007-0447. Epub 2007 Jun 19. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.