

Description
15q11-q13 duplication syndrome (dup15q syndrome) is a developmental disorder; its signs and symptoms vary among affected individuals.
Poor muscle tone (hypotonia) is common in individuals with dup15q syndrome and contributes to delayed development and impairment of motor skills, including sitting and walking. Most affected children develop the ability to walk independently after age 2 or 3, and they typically have a wide-based or uncoordinated (ataxic) pattern of walking (gait). Babies with dup15q syndrome often have trouble feeding due to weak facial muscles that impair sucking and swallowing.
Intellectual disability also occurs in people with dup15q syndrome and can range from mild to profound; however, it is usually in the moderate to severe range. Speech and language development are particularly affected, with some individuals never developing functional speech. Most individuals with this disorder have autism spectrum disorder (ASD), and many have language problems associated with ASD such as repeating the words of others (echolalia) or repeating particular phrases (stereotypical utterances).
Behavioral difficulties are also associated with dup15q syndrome, including other features of ASD such as difficulty with changes in routine and problems with social interaction. Affected individuals may also experience hyperactivity, anxiety, and frustration leading to tantrums. Mood disorders and psychosis occur in some affected individuals.
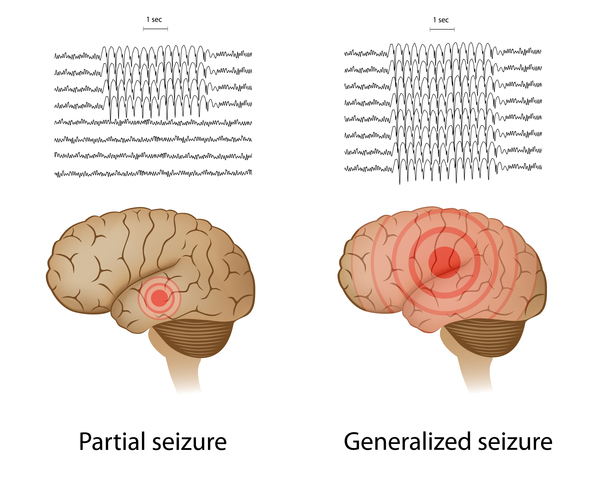
More than half of people with dup15q syndrome have recurrent seizures (epilepsy). The seizures usually develop between the ages of 6 months and 9 years. Some people with dup15q syndrome have only focal seizures, which affect one part of the brain and usually do not cause a loss of consciousness. In other affected individuals, seizures begin with a type called infantile spasms (seizures that usually appear before the age of 1 and involve recurrent muscle contractions) and later include other types of seizures. In addition to focal seizures, these can include rapid uncontrolled muscle jerks (myotonic seizures); tonic-clonic (also called grand mal) seizures, which involve rigidity, convulsions, and loss of consciousness; and absence (also known as petit mal) seizures, which are brief episodes of impaired consciousness that look like staring spells. Affected individuals may develop complex, difficult-to-treat (intractable) seizure patterns such as Lennox-Gastaut syndrome. Seizures can lead to falls, loss of developmental milestones (developmental regression), and in a small minority of cases, sudden death during sleep (called sudden unexpected death in epilepsy, or SUDEP).
Hearing loss in childhood is common in dup15q syndrome and usually results from ear infections that cause fluid buildup in the middle ear. This hearing loss is often temporary. However, if ear infections are left untreated during early childhood, the hearing loss can interfere with language development and worsen the speech problems associated with dup15q syndrome.

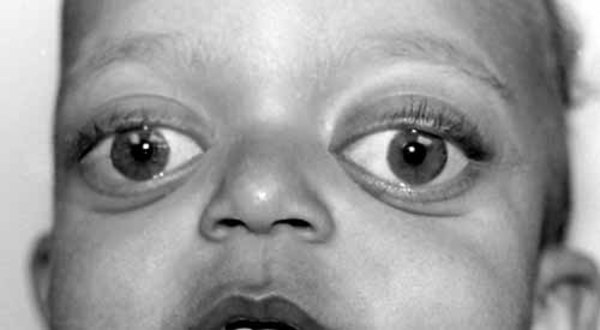
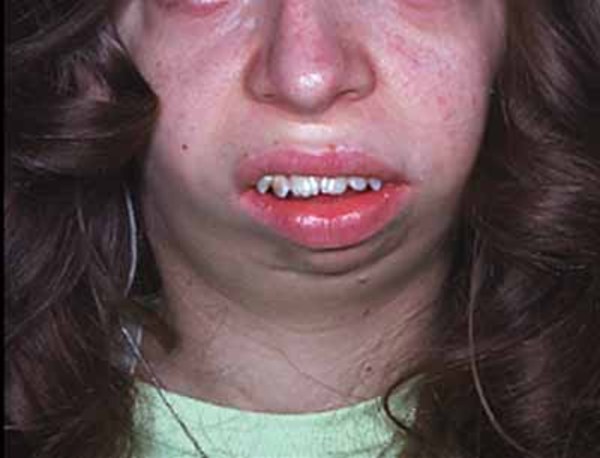
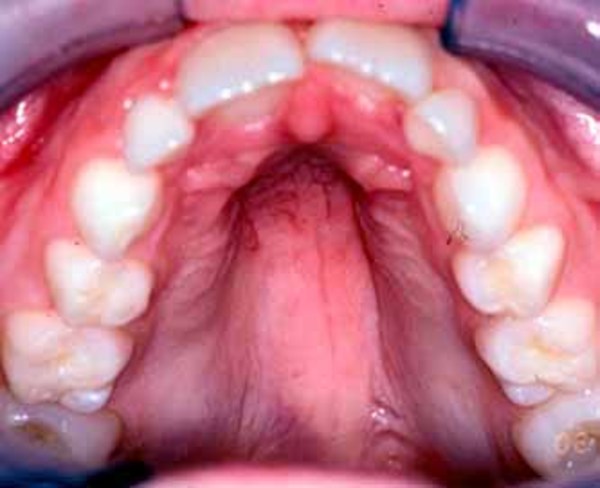
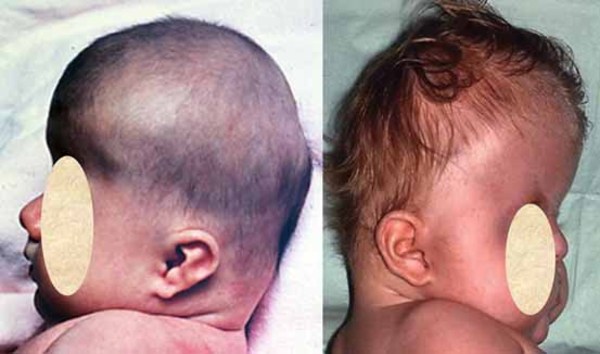
About 30 percent of individuals with dup15q syndrome are born with eyes that do not look in the same direction (strabismus). Other unusual facial features that can occur in this condition include a low forehead; outside corners of the eyes that point downward (downslanting palpebral fissures); a flattened nasal bridge with a short, upturned nose; nostrils that open to the front rather than downward (anteverted nares); a long space between the nose and the upper lip (philtrum); a small lower jaw (micrognathia); a high-arched roof of the mouth (palate); full lips; low-set ears; and a flat back of the head (occiput). These features are typically subtle and may not be noticed during infancy.
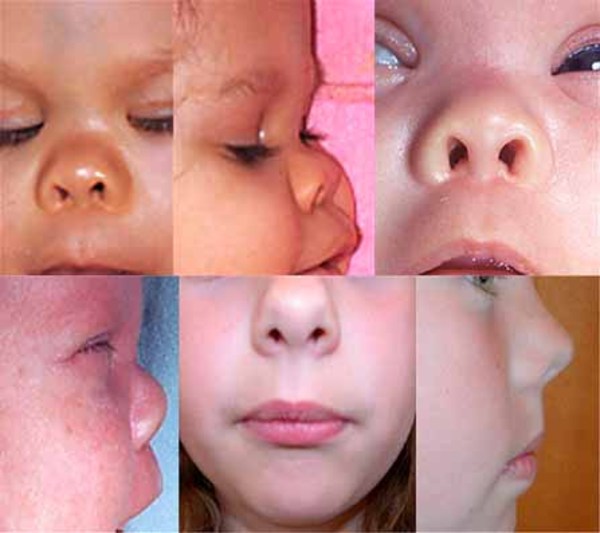
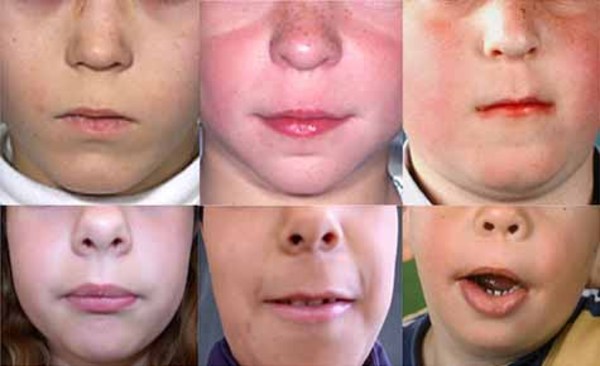
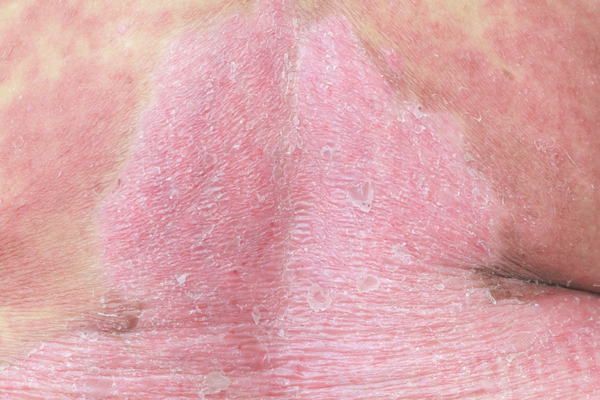
Other problems associated with dup15q syndrome in some affected individuals include a reduced ability to feel pain; a spine that curves to the side (scoliosis); recurrent respiratory infections in childhood; a skin condition called eczema; early (precocious) puberty and, in females, menstrual irregularities; minor genital abnormalities in males such as undescended testes (cryptorchidism); overeating; and excessive weight gain.

Frequency
The prevalence of dup15q syndrome is unknown. It may be as high as 1 in 5,000 individuals in the general population and is thought to be about 10 times more common in people with ASD or intellectual disability.
Causes
Dup15q syndrome is caused by chromosome abnormalities that result in at least one extra copy of a region of chromosome 15 called 15q11.2-q13.1. In particular, the condition arises only if the chromosome abnormality occurs on the copy of the chromosome inherited from the mother (the maternal copy). People normally inherit one copy of chromosome 15 from each parent. However, some genes on this chromosome, including some of those in the 15q11.2-q13.1 region, are turned on (active) only on the maternal copy. This parent-specific gene activation results from a phenomenon called genomic imprinting.
The most common chromosome abnormality that leads to 15q11.2-q13.1 duplication, occurring in about 80 percent of people with dup15q syndrome, is called an isodicentric chromosome 15. An isodicentric chromosome contains mirror-image segments of genetic material and has two constriction points (centromeres), rather than one centromere as in normal chromosomes. In people with an isodicentric chromosome 15, cells have the usual two copies of chromosome 15 plus the two duplicated copies of the segment of genetic material in the isodicentric chromosome, for a total of four copies of the duplicated segment.
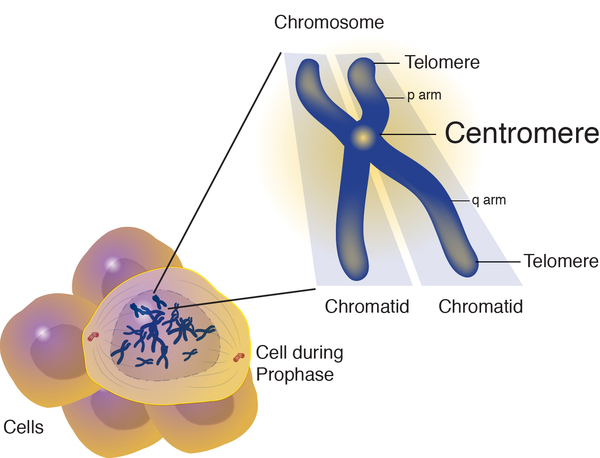
In about 20 percent of cases of dup15q syndrome, the duplication occurs on the long (q) arm of one of the two copies of chromosome 15 in each cell; this situation is called an interstitial duplication. In these cases, cells have two copies of chromosome 15, one of which has an extra copy of the segment of genetic material, for a total of three copies of the duplicated segment.
In all cases of dup15q syndrome, the duplicated genetic material results in extra copies of certain genes involved in development. This extra genetic material disrupts normal development, causing the characteristic features of this disorder. People with dup15q syndrome resulting from an interstitial duplication often have milder signs and symptoms than those in whom the disorder results from an isodicentric chromosome 15.
Inheritance
Dup15q syndrome caused by an isodicentric chromosome 15 is usually not inherited. The chromosomal change that causes the disorder is typically de novo, which means it occurs as a random event during the formation of eggs in the mother of the affected individual. Most affected individuals have no history of the disorder in their family.
In 15 percent of cases of dup15q syndrome caused by an interstitial duplication, the condition is inherited from a mother who also has the duplication. The remainder of cases are caused by de novo duplication of the genetic material.
Other Names for This Condition
- Dup15q syndrome
- Duplication/inversion 15q11
- Idic(15)
- Inv dup(15)
- Inverted duplication 15
- Isodicentric chromosome 15
- Isodicentric chromosome 15 syndrome
- Non-distal tetrasomy 15q
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Battaglia A, Bernardini L, Torrente I, Novelli A, Scarselli G. Spectrum of epilepsy and electroencephalogram patterns in idic (15) syndrome. Am J Med Genet A. 2016 Oct;170(10):2531-9. doi: 10.1002/ajmg.a.37844. Epub 2016 Aug 11. Citation on PubMed
- Conant KD, Finucane B, Cleary N, Martin A, Muss C, Delany M, Murphy EK, Rabe O, Luchsinger K, Spence SJ, Schanen C, Devinsky O, Cook EH, LaSalle J, Reiter LT, Thibert RL. A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia. 2014 Mar;55(3):396-402. doi: 10.1111/epi.12530. Epub 2014 Feb 6. Citation on PubMed
- DiStefano C, Gulsrud A, Huberty S, Kasari C, Cook E, Reiter LT, Thibert R, Jeste SS. Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J Neurodev Disord. 2016 May 6;8:19. doi: 10.1186/s11689-016-9152-y. eCollection 2016. Citation on PubMed or Free article on PubMed Central
- Friedman D, Thaler A, Thaler J, Rai S, Cook E, Schanen C, Devinsky O. Mortality in isodicentric chromosome 15 syndrome: The role of SUDEP. Epilepsy Behav. 2016 Aug;61:1-5. doi: 10.1016/j.yebeh.2016.04.001. Epub 2016 May 21. Citation on PubMed
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010 May;38(2):181-91. doi: 10.1016/j.nbd.2008.08.011. Epub 2008 Sep 18. Citation on PubMed or Free article on PubMed Central
- Isles AR, Ingason A, Lowther C, Walters J, Gawlick M, Stober G, Rees E, Martin J, Little RB, Potter H, Georgieva L, Pizzo L, Ozaki N, Aleksic B, Kushima I, Ikeda M, Iwata N, Levinson DF, Gejman PV, Shi J, Sanders AR, Duan J, Willis J, Sisodiya S, Costain G, Werge TM, Degenhardt F, Giegling I, Rujescu D, Hreidarsson SJ, Saemundsen E, Ahn JW, Ogilvie C, Girirajan SD, Stefansson H, Stefansson K, O'Donovan MC, Owen MJ, Bassett A, Kirov G. Parental Origin of Interstitial Duplications at 15q11.2-q13.3 in Schizophrenia and Neurodevelopmental Disorders. PLoS Genet. 2016 May 6;12(5):e1005993. doi: 10.1371/journal.pgen.1005993. eCollection 2016 May. Citation on PubMed or Free article on PubMed Central
- Kwasnicka-Crawford DA, Roberts W, Scherer SW. Characterization of an autism-associated segmental maternal heterodisomy of the chromosome 15q11-13 region. J Autism Dev Disord. 2007 Apr;37(4):694-702. doi: 10.1007/s10803-006-0225-8. Citation on PubMed
- Luchsinger K, Lau H, Hedlund JL, Friedman D, Krushel K, Devinsky O. Parental-reported pain insensitivity in Dup15q. Epilepsy Behav. 2016 Feb;55:124-7. doi: 10.1016/j.yebeh.2015.10.007. Epub 2016 Jan 13. Citation on PubMed
- Lusk L, Vogel-Farley V, DiStefano C, Jeste S. Maternal 15q Duplication Syndrome. 2016 Jun 16 [updated 2021 Jul 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK367946/ Citation on PubMed
- Matricardi S, Darra F, Spalice A, Basti C, Fontana E, Dalla Bernardina B, Elia M, Giordano L, Accorsi P, Cusmai R, De Liso P, Romeo A, Ragona F, Granata T, Concolino D, Carotenuto M, Pavone P, Pruna D, Striano P, Savasta S, Verrotti A. Electroclinical findings and long-term outcomes in epileptic patients with inv dup (15). Acta Neurol Scand. 2018 Jun;137(6):575-581. doi: 10.1111/ane.12902. Epub 2018 Jan 23. Citation on PubMed
- Verrotti A, Sertorio F, Matricardi S, Ferrara P, Striano P. Electroclinical features of epilepsy in patients with InvDup(15). Seizure. 2017 Apr;47:87-91. doi: 10.1016/j.seizure.2017.03.006. Epub 2017 Mar 9. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.