

Description
X-linked myotubular myopathy is a condition that primarily affects muscles used for movement (skeletal muscles) and occurs almost exclusively in males. People with this condition have muscle weakness (myopathy) and decreased muscle tone (hypotonia) that are usually evident at birth. When viewed under a microscope, the muscle fibers of affected individuals are typically small and underdeveloped.
The muscle problems in X-linked myotubular myopathy impair the development of motor skills such as sitting, standing, and walking. Affected infants may also have difficulties with feeding due to muscle weakness. Individuals with this condition often do not have the muscle strength to breathe regularly on their own and must be supported with a machine to help them get enough oxygen (mechanical ventilation). Some affected individuals need breathing assistance only periodically, typically during sleep, while others require it continuously. People with X-linked myotubular myopathy may also have weakness in the muscles that control eye movement (ophthalmoplegia), weakness in other muscles of the face, and absent reflexes (areflexia).
In X-linked myotubular myopathy, muscle weakness often disrupts normal bone development and can lead to fragile bones, an abnormal curvature of the spine (scoliosis), and joint deformities (contractures) of the hips and knees. People with X-linked myotubular myopathy may have a large head with a narrow and elongated face and a high, arched roof of the mouth (palate). They may also have recurrent ear and respiratory infections, seizures, or liver disease. Some affected individuals develop a serious liver condition called peliosis hepatitis, which can cause life-threatening bleeding (hemorrhage).

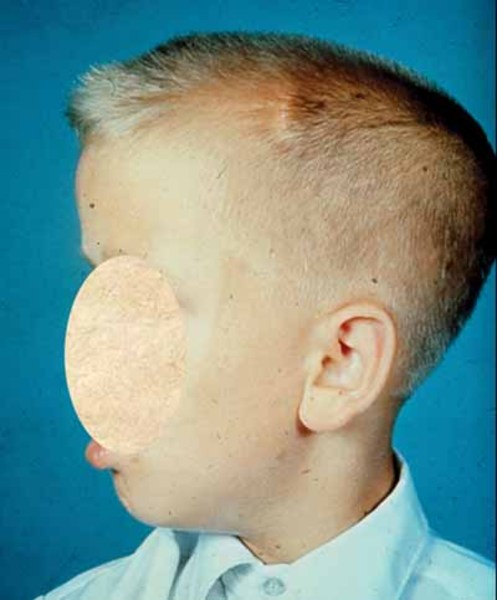
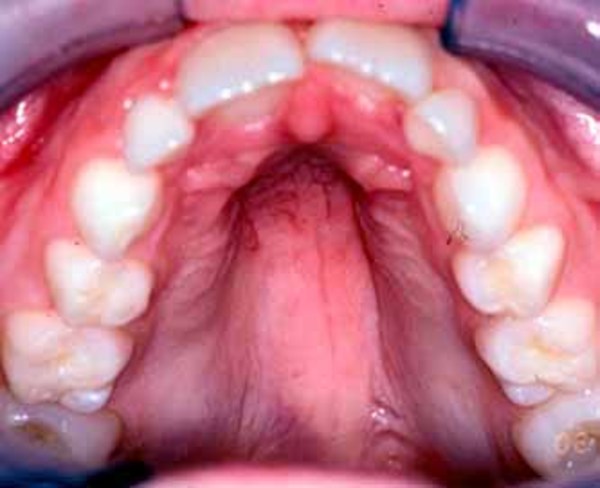
Because of their severe breathing problems, individuals with X-linked myotubular myopathy usually survive only into early childhood; however, some people with this condition have lived into adulthood.
X-linked myotubular myopathy is the most severe condition in a group of disorders called centronuclear myopathy. In centronuclear myopathy, the nucleus is found at the center of many rod-shaped muscle cells instead of at either end, where it is normally located.
Frequency
The incidence of X-linked myotubular myopathy is estimated to be 1 in 50,000 newborn males worldwide.
Causes
Mutations in the MTM1 gene cause X-linked myotubular myopathy. The MTM1 gene provides instructions for producing an enzyme called myotubularin. Myotubularin is thought to be involved in the development and maintenance of muscle cells.
MTM1 gene mutations probably disrupt myotubularin's role in muscle cell development and maintenance, causing muscle weakness and other signs and symptoms of X-linked myotubular myopathy.
Inheritance
X-linked myotubular myopathy is inherited in an X-linked recessive pattern . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In X-linked myotubular myopathy, the affected male inherits one altered copy from his mother in 80 to 90 percent of cases. In the remaining 10 to 20 percent of cases, the disorder results from a new mutation in the gene that occurs during the formation of a parent's reproductive cells (eggs or sperm) or in early embryonic development. Females with one altered copy of the MTM1 gene generally do not experience signs and symptoms of the disorder. In some cases, however, females who have one altered copy of the MTM1 gene experience mild signs and symptoms of X-linked myotubular myopathy, including changes in the muscle fibers, muscle weakness, and breathing problems. Researchers are studying why some females are affected and others are not.
Other Names for This Condition
- CNM
- MTMX
- X-linked centronuclear myopathy
- XLMTM
- XMTM
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G, D'Apice MR, Lagier-Tourenne C, Buj-Bello A, Romero NB, Mandel JL. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. 2003 Feb;112(2):135-42. doi: 10.1007/s00439-002-0869-1. Epub 2002 Nov 28. Citation on PubMed
- Biancalana V, Scheidecker S, Miguet M, Laquerriere A, Romero NB, Stojkovic T, Abath Neto O, Mercier S, Voermans N, Tanner L, Rogers C, Ollagnon-Roman E, Roper H, Boutte C, Ben-Shachar S, Lornage X, Vasli N, Schaefer E, Laforet P, Pouget J, Moerman A, Pasquier L, Marcorelle P, Magot A, Kusters B, Streichenberger N, Tranchant C, Dondaine N, Schneider R, Gasnier C, Calmels N, Kremer V, Nguyen K, Perrier J, Kamsteeg EJ, Carlier P, Carlier RY, Thompson J, Boland A, Deleuze JF, Fardeau M, Zanoteli E, Eymard B, Laporte J. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues. Acta Neuropathol. 2017 Dec;134(6):889-904. doi: 10.1007/s00401-017-1748-0. Epub 2017 Jul 6. Citation on PubMed
- Bijarnia S, Puri RD, Jain M, Kler N, Roy S, Urtizberea JA, Biancalana V, Verma IC. Mutation studies in X-linked myotubular myopathy in three Indian families. Indian J Pediatr. 2010 Apr;77(4):431-3. doi: 10.1007/s12098-010-0057-6. Epub 2010 Mar 31. Citation on PubMed
- Cahill PJ, Rinella AS, Bielski RJ. Orthopaedic complications of myotubular myopathy. J Pediatr Orthop. 2007 Jan-Feb;27(1):98-103. doi: 10.1097/BPO.0b013e31802b6c73. Citation on PubMed
- Dowling JJ, Lawlor MW, Das S. X-Linked Myotubular Myopathy. 2002 Feb 25 [updated 2018 Aug 23]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1432/ Citation on PubMed
- Echaniz-Laguna A, Biancalana V, Bohm J, Tranchant C, Mandel JL, Laporte J. Adult centronuclear myopathies: A hospital-based study. Rev Neurol (Paris). 2013 Aug-Sep;169(8-9):625-31. doi: 10.1016/j.neurol.2012.12.006. Epub 2013 Aug 9. Citation on PubMed
- Hoffjan S, Thiels C, Vorgerd M, Neuen-Jacob E, Epplen JT, Kress W. Extreme phenotypic variability in a German family with X-linked myotubular myopathy associated with E404K mutation in MTM1. Neuromuscul Disord. 2006 Nov;16(11):749-53. doi: 10.1016/j.nmd.2006.07.020. Epub 2006 Sep 26. Citation on PubMed
- Lawlor MW, Alexander MS, Viola MG, Meng H, Joubert R, Gupta V, Motohashi N, Manfready RA, Hsu CP, Huang P, Buj-Bello A, Kunkel LM, Beggs AH, Gussoni E. Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am J Pathol. 2012 Sep;181(3):961-8. doi: 10.1016/j.ajpath.2012.05.016. Epub 2012 Jul 27. Citation on PubMed or Free article on PubMed Central
- McEntagart M, Parsons G, Buj-Bello A, Biancalana V, Fenton I, Little M, Krawczak M, Thomas N, Herman G, Clarke A, Wallgren-Pettersson C. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. 2002 Dec;12(10):939-46. doi: 10.1016/s0960-8966(02)00153-0. Citation on PubMed
- Pierson CR, Tomczak K, Agrawal P, Moghadaszadeh B, Beggs AH. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005 Jul;64(7):555-64. doi: 10.1097/01.jnen.0000171653.17213.2e. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.