

Description
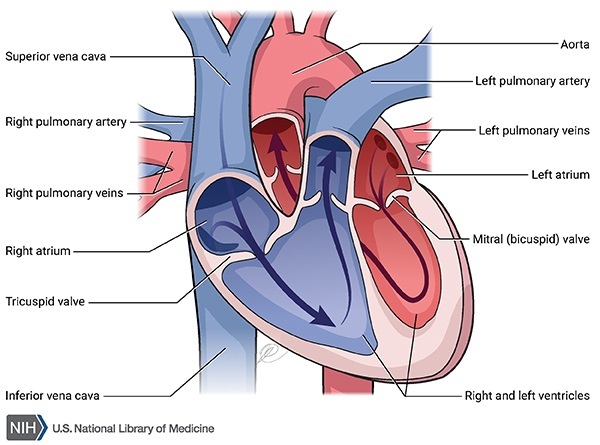
X-linked dilated cardiomyopathy is a form of heart disease. Dilated cardiomyopathy enlarges and weakens the heart (cardiac) muscle, preventing the heart from pumping blood efficiently. Signs and symptoms of this condition can include an irregular heartbeat (arrhythmia), shortness of breath, extreme tiredness (fatigue), and swelling of the legs and feet. In males with X-linked dilated cardiomyopathy, heart problems usually develop early in life and worsen quickly, leading to heart failure in adolescence or early adulthood. In affected females, the condition appears later in life and worsens more slowly.
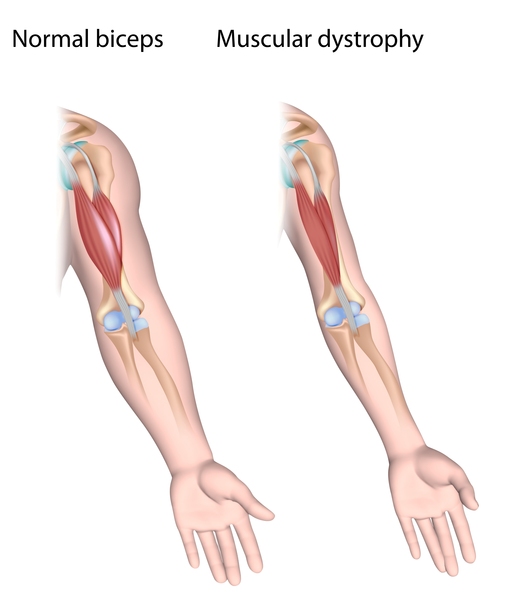
X-linked dilated cardiomyopathy is part of a spectrum of related conditions caused by mutations in the DMD gene. The other conditions in the spectrum, Duchenne and Becker muscular dystrophy, are characterized by progressive weakness and wasting of muscles used for movement (skeletal muscles) in addition to heart disease. People with X-linked dilated cardiomyopathy typically do not have any skeletal muscle weakness or wasting, although they may have subtle changes in their skeletal muscle cells that are detectable through laboratory testing. Based on these skeletal muscle changes, X-linked dilated cardiomyopathy is sometimes classified as subclinical Becker muscular dystrophy.
Frequency
X-linked dilated cardiomyopathy appears to be an uncommon condition, although its prevalence is unknown.
Causes
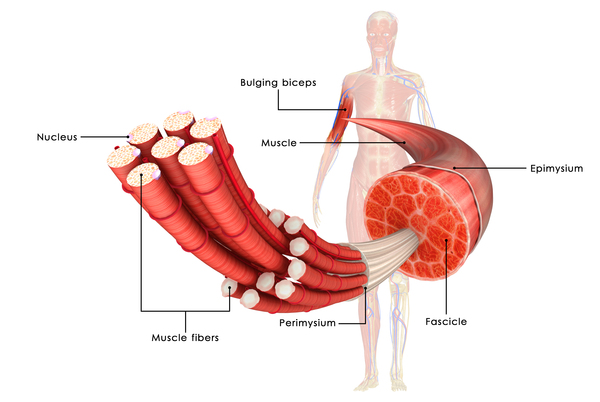
X-linked dilated cardiomyopathy results from mutations in the DMD gene. This gene provides instructions for making a protein called dystrophin, which helps stabilize and protect muscle fibers and may play a role in chemical signaling within cells. The mutations responsible for X-linked dilated cardiomyopathy preferentially affect the activity of dystrophin in cardiac muscle cells. As a result of these mutations, affected individuals typically have little or no functional dystrophin in the heart. Without enough of this protein, cardiac muscle cells become damaged as the heart muscle repeatedly contracts and relaxes. The damaged muscle cells weaken and die over time, leading to the heart problems characteristic of X-linked dilated cardiomyopathy.
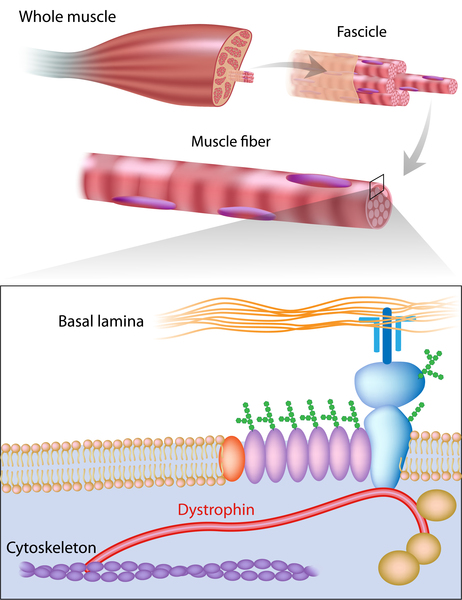
The mutations that cause X-linked dilated cardiomyopathy often lead to reduced amounts of dystrophin in skeletal muscle cells. However, enough of this protein is present to prevent weakness and wasting of the skeletal muscles.
Because X-linked dilated cardiomyopathy results from a shortage of dystrophin, it is classified as a dystrophinopathy.
Inheritance
As its name suggests, X-linked dilated cardiomyopathy has an X-linked pattern of inheritance. The DMD gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell usually leads to relatively mild heart disease that appears later in life. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes more severe signs and symptoms that occur earlier in life. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- CMD3B
- Dilated cardiomyopathy 3B
- DMD-associated dilated cardiomyopathy
- DMD-related dilated cardiomyopathy
- XLCM
- XLDC
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Beggs AH. Dystrophinopathy, the expanding phenotype. Dystrophin abnormalities in X-linked dilated cardiomyopathy. Circulation. 1997 May 20;95(10):2344-7. doi: 10.1161/01.cir.95.10.2344. No abstract available. Citation on PubMed
- Berko BA, Swift M. X-linked dilated cardiomyopathy. N Engl J Med. 1987 May 7;316(19):1186-91. doi: 10.1056/NEJM198705073161904. Citation on PubMed
- Cohen N, Muntoni F. Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy. Heart. 2004 Aug;90(8):835-41. doi: 10.1136/hrt.2003.023390. Citation on PubMed or Free article on PubMed Central
- Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. 2000 Sep 5 [updated 2022 Jan 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1119/ Citation on PubMed
- Ferlini A, Sewry C, Melis MA, Mateddu A, Muntoni F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul Disord. 1999 Jul;9(5):339-46. doi: 10.1016/s0960-8966(99)00015-2. Citation on PubMed
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003 Dec;2(12):731-40. doi: 10.1016/s1474-4422(03)00585-4. Citation on PubMed
- Nakamura A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel). 2015 Jun 9;8(2):303-20. doi: 10.3390/ph8020303. Citation on PubMed or Free article on PubMed Central
- Neri M, Valli E, Alfano G, Bovolenta M, Spitali P, Rapezzi C, Muntoni F, Banfi S, Perini G, Gualandi F, Ferlini A. The absence of dystrophin brain isoform expression in healthy human heart ventricles explains the pathogenesis of 5' X-linked dilated cardiomyopathy. BMC Med Genet. 2012 Mar 28;13:20. doi: 10.1186/1471-2350-13-20. Citation on PubMed or Free article on PubMed Central
- Posafalvi A, Herkert JC, Sinke RJ, van den Berg MP, Mogensen J, Jongbloed JD, van Tintelen JP. Clinical utility gene card for: dilated cardiomyopathy (CMD). Eur J Hum Genet. 2013 Oct;21(10). doi: 10.1038/ejhg.2012.276. Epub 2012 Dec 19. No abstract available. Citation on PubMed or Free article on PubMed Central
- Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993 Jun;87(6):1854-65. doi: 10.1161/01.cir.87.6.1854. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.