

Description
X-linked chondrodysplasia punctata 2 is a disorder characterized by bone, skin, and eye abnormalities. It occurs almost exclusively in females.
Although the signs and symptoms of this condition vary widely, almost all affected individuals have chondrodysplasia punctata, an abnormality that appears on x-rays as spots (stippling) near the ends of bones and in cartilage. In this form of chondrodysplasia punctata, the stippling typically affects the long bones in the arms and legs, the ribs, the spinal bones (vertebrae), and the cartilage that makes up the windpipe (trachea). The stippling is apparent in infancy but disappears in early childhood. Other skeletal abnormalities seen in people with X-linked chondrodysplasia punctata 2 include shortening of the bones in the upper arms and thighs (rhizomelia) that is often different on the right and left sides, and progressive abnormal curvature of the spine (kyphoscoliosis). As a result of these abnormalities, people with this condition tend to have short stature.
Infants with X-linked chondrodysplasia punctata 2 are born with dry, scaly patches of skin (ichthyosis) in a linear or spiral (whorled) pattern. The scaly patches fade over time, leaving abnormally colored blotches of skin without hair (follicular atrophoderma). Most affected individuals also have sparse, coarse hair on their scalps.
Most people with X-linked chondrodysplasia punctata 2 have clouding of the lens of the eye (cataracts) from birth or early childhood. Other eye abnormalities that have been associated with this disorder include unusually small eyes (microphthalmia) and small corneas (microcornea). The cornea is the clear front surface of the eye. These eye abnormalities can impair vision.

In affected females, X-linked chondrodysplasia punctata 2 is typically associated with normal intelligence and a normal lifespan. However, a much more severe form of the condition has been reported in a small number of males. Affected males have some of the same features as affected females, as well as weak muscle tone (hypotonia), changes in the structure of the brain, moderately to profoundly delayed development, seizures, distinctive facial features, and other birth defects. The health problems associated with X-linked chondrodysplasia punctata 2 are often life-threatening in males.
Frequency
X-linked chondrodysplasia punctata 2 has been estimated to affect fewer than 1 in 400,000 newborns. However, the disorder may actually be more common than this estimate because it is likely underdiagnosed, particularly in females with mild signs and symptoms.
More than 95 percent of cases of X-linked chondrodysplasia punctata 2 occur in females. About a dozen males with the condition have been reported in the scientific literature.
Causes
X-linked chondrodysplasia punctata 2 is caused by mutations in the EBP gene. This gene provides instructions for making an enzyme called 3β-hydroxysteroid-Δ8,Δ7-isomerase, which is responsible for one of the final steps in the production of cholesterol. Cholesterol is a waxy, fat-like substance that is produced in the body and obtained from foods that come from animals (particularly egg yolks, meat, poultry, fish, and dairy products). Although too much cholesterol is a risk factor for heart disease, this molecule is necessary for normal embryonic development and has important functions both before and after birth. It is a structural component of cell membranes and plays a role in the production of certain hormones and digestive acids.
Mutations in the EBP gene reduce the activity of 3β-hydroxysteroid-Δ8,Δ7-isomerase, preventing cells from producing enough cholesterol. A shortage of this enzyme also allows potentially toxic byproducts of cholesterol production to build up in the body. The combination of low cholesterol levels and an accumulation of other substances likely disrupts the growth and development of many body systems. It is not known, however, how this disturbance in cholesterol production leads to the specific features of X-linked chondrodysplasia punctata 2.
Inheritance
This condition is inherited in an X-linked dominant pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the EBP gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of 3β-hydroxysteroid-Δ8,Δ7-isomerase and other cells produce none. The resulting overall reduction in the amount of this enzyme underlies the signs and symptoms of X-linked chondrodysplasia punctata 2.
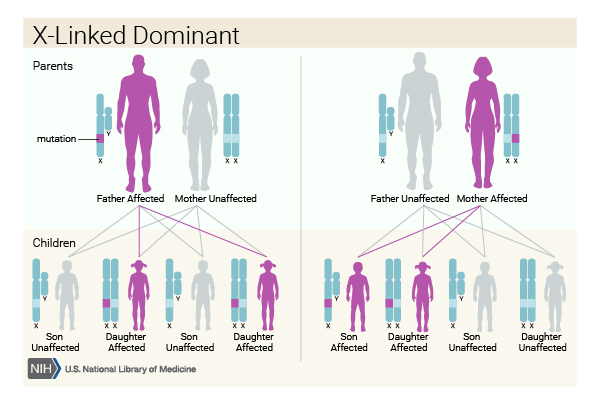

In males (who have only one X chromosome), a mutation in the EBP gene can result in a total loss of 3β-hydroxysteroid-Δ8,Δ7-isomerase. A complete lack of this enzyme is usually lethal in the early stages of development, so few males have been born with X-linked chondrodysplasia punctata 2.
Other Names for This Condition
- CDPX2
- Chondrodysplasia punctata 2, X-linked
- Conradi-Hünermann syndrome
- Conradi-Hünermann-Happle syndrome
- Happle syndrome
- X-linked dominant chondrodysplasia punctata
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aughton DJ, Kelley RI, Metzenberg A, Pureza V, Pauli RM. X-linked dominant chondrodysplasia punctata (CDPX2) caused by single gene mosaicism in a male. Am J Med Genet A. 2003 Jan 30;116A(3):255-60. doi: 10.1002/ajmg.a.10852. Citation on PubMed
- Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L, Kelley RI, Valle D. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999 Jul;22(3):291-4. doi: 10.1038/10357. Citation on PubMed
- Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, Boyd Y, Herman GE. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. jderry@immunex.com. Nat Genet. 1999 Jul;22(3):286-90. doi: 10.1038/10350. Citation on PubMed
- Furtado LV, Bayrak-Toydemir P, Hulinsky B, Damjanovich K, Carey JC, Rope AF. A novel X-linked multiple congenital anomaly syndrome associated with an EBP mutation. Am J Med Genet A. 2010 Nov;152A(11):2838-44. doi: 10.1002/ajmg.a.33674. Citation on PubMed
- Herman GE, Kelley RI, Pureza V, Smith D, Kopacz K, Pitt J, Sutphen R, Sheffield LJ, Metzenberg AB. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome). Genet Med. 2002 Nov-Dec;4(6):434-8. doi: 10.1097/00125817-200211000-00006. Citation on PubMed
- Hoang MP, Carder KR, Pandya AG, Bennett MJ. Ichthyosis and keratotic follicular plugs containing dystrophic calcification in newborns: distinctive histopathologic features of x-linked dominant chondrodysplasia punctata (Conradi-Hunermann-Happle syndrome). Am J Dermatopathol. 2004 Feb;26(1):53-8. doi: 10.1097/00000372-200402000-00007. Citation on PubMed
- Ikegawa S, Ohashi H, Ogata T, Honda A, Tsukahara M, Kubo T, Kimizuka M, Shimode M, Hasegawa T, Nishimura G, Nakamura Y. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet. 2000 Oct 2;94(4):300-5. doi: 10.1002/1096-8628(20001002)94:43.0.co;2-3. Citation on PubMed
- Kumble S, Savarirayan R. Chondrodysplasia Punctata 2, X-Linked. 2011 May 31 [updated 2020 Jan 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK55062/ Citation on PubMed
- Milunsky JM, Maher TA, Metzenberg AB. Molecular, biochemical, and phenotypic analysis of a hemizygous male with a severe atypical phenotype for X-linked dominant Conradi-Hunermann-Happle syndrome and a mutation in EBP. Am J Med Genet A. 2003 Jan 30;116A(3):249-54. doi: 10.1002/ajmg.a.10849. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.