

Description
Wolf-Hirschhorn syndrome is a condition that affects many parts of the body. The major features of this disorder include a characteristic facial features, delayed growth and development, intellectual disability, and seizures.
Almost everyone with this disorder has distinctive facial features, including a broad nasal bridge, large and protruding eyes, and a high forehead. This combination is described as a "Greek warrior helmet" appearance. Other characteristic facial features include a shortened distance between the nose and upper lip (a short philtrum), a downturned mouth, a small chin (micrognathia), and poorly formed ears with small holes (pits) or flaps of skin (tags). Additionally, affected individuals may have asymmetrical facial features and an unusually small head (microcephaly ).
).
People with Wolf-Hirschhorn syndrome experience delayed growth and development. Slow growth begins before birth, and affected infants tend to have problems feeding and gaining weight (failure to thrive). They also have weak muscle tone (hypotonia) and underdeveloped muscles. Motor skills such as sitting, standing, and walking are significantly delayed. Most children and adults with this disorder also have short stature.
Intellectual disability ranges from mild to severe in people with Wolf-Hirschhorn syndrome. Compared to people with other forms of intellectual disability, their socialization skills are strong, but verbal communication and language skills tend to be weaker. Most affected children also have seizures, which may be resistant to treatment. Seizures tend to disappear with age.
Additional features of Wolf-Hirschhorn syndrome include skin changes, such as mottled or dry skin; skeletal abnormalities, such as abnormal curvature of the spine (scoliosis and kyphosis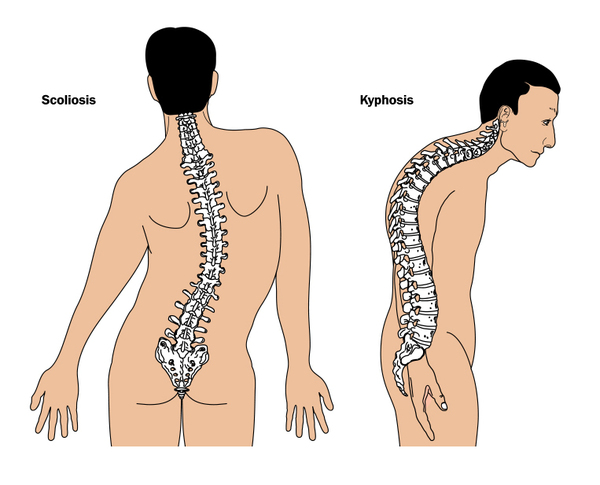 ); dental problems including, missing teeth; and an opening in the roof of the mouth (cleft palate
); dental problems including, missing teeth; and an opening in the roof of the mouth (cleft palate ) and/or a split in the upper lip (cleft lip
) and/or a split in the upper lip (cleft lip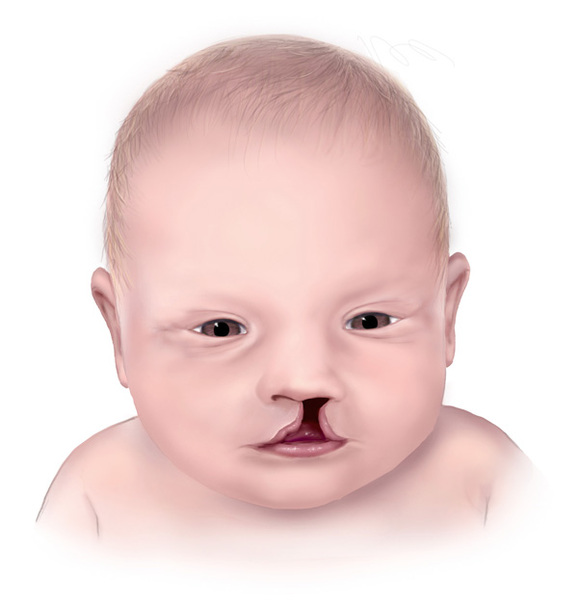 ). Wolf-Hirschhorn syndrome can also cause abnormalities of the eyes, heart, and genitourinary tract.
). Wolf-Hirschhorn syndrome can also cause abnormalities of the eyes, heart, and genitourinary tract.
A condition called Pitt-Rogers-Danks syndrome has features that overlap with those of Wolf-Hirschhorn syndrome. Researchers now recognize that these two conditions are actually part of a single syndrome with variable signs and symptoms.
Frequency
The prevalence of Wolf-Hirschhorn syndrome is estimated to be 1 in 50,000 births. However, it is likely that some affected individuals are never diagnosed.
For unknown reasons, Wolf-Hirschhorn syndrome occurs in about twice as many females as males.
Causes
Wolf-Hirschhorn syndrome is caused by a deletion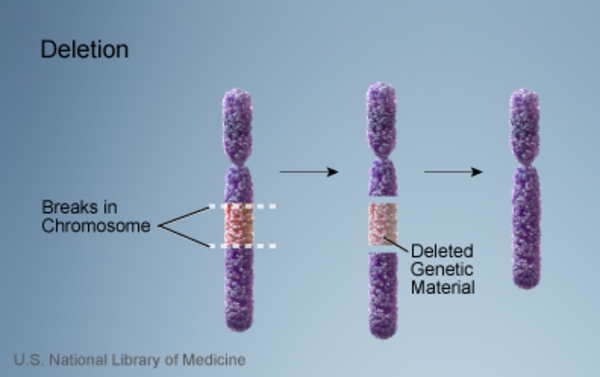 of genetic material near the end of the short (p) arm of chromosome 4. This chromosomal change is sometimes written as 4p-. The size of the deletion varies among affected individuals; studies suggest that larger deletions tend to result in more severe intellectual disability and physical abnormalities than smaller deletions.
of genetic material near the end of the short (p) arm of chromosome 4. This chromosomal change is sometimes written as 4p-. The size of the deletion varies among affected individuals; studies suggest that larger deletions tend to result in more severe intellectual disability and physical abnormalities than smaller deletions.
The signs and symptoms of Wolf-Hirschhorn are related to the loss of multiple genes on the short arm of chromosome 4. NSD2, LETM1, and MSX1 are the genes that are deleted in people with the typical signs and symptoms of this disorder. These genes play significant roles in early development, although many of their specific functions are unknown.
Researchers believe that the loss of the NSD2 gene can cause many of the characteristic features of Wolf-Hirschhorn syndrome, including the distinctive facial appearance and developmental delay. The deletion of the LETM1 gene and other nearby genes (including CPLX1) may cause seizures or other abnormal electrical activity in the brain. The loss of the MSX1 gene may be responsible for the dental abnormalities and cleft lip and/or palate that are often seen in people with this condition.
Scientists are working to identify additional genes at the end of the short arm of chromosome 4 that contribute to the characteristic features of Wolf-Hirschhorn syndrome.
Inheritance
Between 85 and 90 percent of all cases of Wolf-Hirschhorn syndrome are not inherited. They are caused by a chromosomal deletion that occurs as a random (de novo) event during the formation of reproductive cells (eggs or sperm) or during early embryonic development. More complex chromosomal rearrangements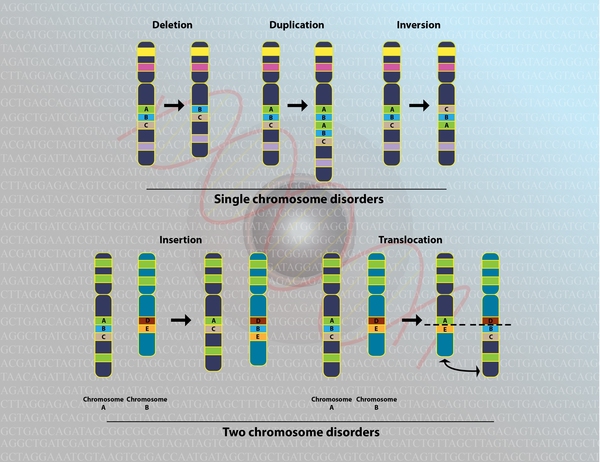 can also occur as de novo events, which may help explain the variability in the condition's signs and symptoms. De novo chromosomal changes occur in people with no history of the disorder in their family.
can also occur as de novo events, which may help explain the variability in the condition's signs and symptoms. De novo chromosomal changes occur in people with no history of the disorder in their family.
An unusual chromosomal abnormality called a ring chromosome can also cause Wolf-Hirschhorn syndrome. Ring chromosomes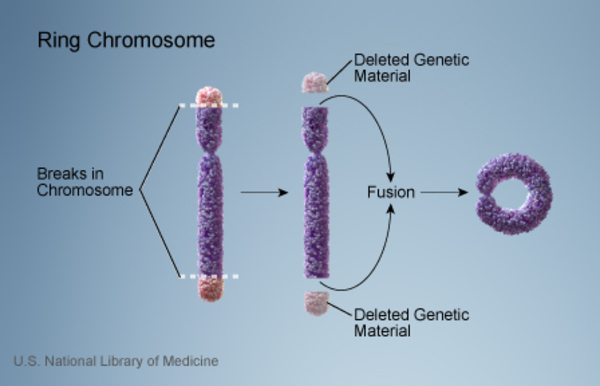 occur when a chromosome breaks in two places and the ends of the chromosome arms fuse together to form a circular structure. In the process, genes near the ends of the chromosome are lost. In a small percentage of people with Wolf-Hirschhorn syndrome, the disorder is caused by chromosome 4 becoming a ring chromosome.
occur when a chromosome breaks in two places and the ends of the chromosome arms fuse together to form a circular structure. In the process, genes near the ends of the chromosome are lost. In a small percentage of people with Wolf-Hirschhorn syndrome, the disorder is caused by chromosome 4 becoming a ring chromosome.
In the remaining cases of Wolf-Hirschhorn syndrome, an affected individual inherits a copy of chromosome 4 with a deleted segment. In these cases, one of the individual's parents carries a balanced translocation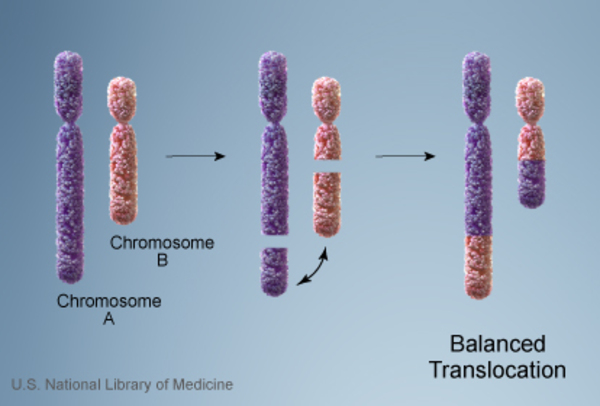 , which occurs when chromosome 4 trades genetic information with another chromosome. No genetic material is gained or lost in a balanced translocation, so these chromosomal changes usually do not cause any health problems. However, translocations can become unbalanced as they are passed to the next generation. Some people with Wolf-Hirschhorn syndrome inherit an unbalanced translocation
, which occurs when chromosome 4 trades genetic information with another chromosome. No genetic material is gained or lost in a balanced translocation, so these chromosomal changes usually do not cause any health problems. However, translocations can become unbalanced as they are passed to the next generation. Some people with Wolf-Hirschhorn syndrome inherit an unbalanced translocation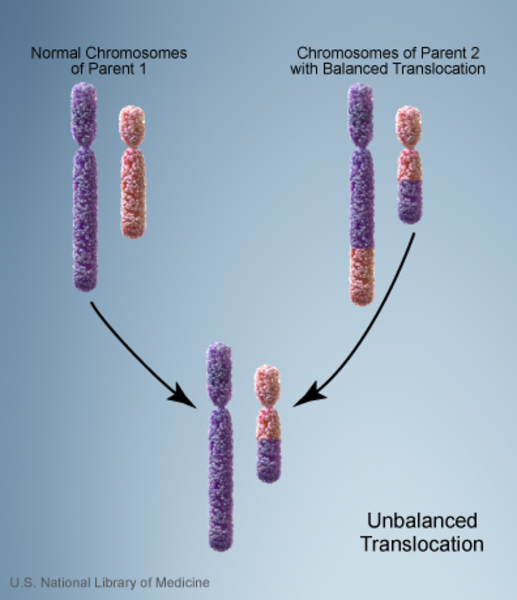 that deletes genes near the end of the short arm of chromosome 4. A loss of these genes results in the intellectual disability, slow growth, and other health problems that are characteristic of this disorder.
that deletes genes near the end of the short arm of chromosome 4. A loss of these genes results in the intellectual disability, slow growth, and other health problems that are characteristic of this disorder.
Other Names for This Condition
- 4p deletion syndrome
- 4p- syndrome
- Chromosome 4p deletion syndrome
- Chromosome 4p monosomy
- Del(4p) syndrome
- Monosomy 4p
- Partial monosomy 4p
- WHS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Battaglia A, Carey JC. Wolf-Hirschhorn syndrome and Pitt-Rogers-Danks syndrome. Am J Med Genet. 1998 Feb 17;75(5):541. doi: 10.1002/(sici)1096-8628(19980217)75:53.0.co;2-k. No abstract available. Citation on PubMed
- Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008 Nov 15;148C(4):246-51. doi: 10.1002/ajmg.c.30187. Citation on PubMed
- Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005 Mar;21(3):188-95. doi: 10.1016/j.tig.2005.01.008. Citation on PubMed
- Fisch GS, Battaglia A, Parrini B, Youngblom J, Simensen R. Cognitive-behavioral features of children with Wolf-Hirschhorn syndrome: preliminary report of 12 cases. Am J Med Genet C Semin Med Genet. 2008 Nov 15;148C(4):252-6. doi: 10.1002/ajmg.c.30185. Citation on PubMed
- Nieminen P, Kotilainen J, Aalto Y, Knuutila S, Pirinen S, Thesleff I. MSX1 gene is deleted in Wolf-Hirschhorn syndrome patients with oligodontia. J Dent Res. 2003 Dec;82(12):1013-7. doi: 10.1177/154405910308201215. Citation on PubMed
- South ST, Hannes F, Fisch GS, Vermeesch JR, Zollino M. Pathogenic significance of deletions distal to the currently described Wolf-Hirschhorn syndrome critical regions on 4p16.3. Am J Med Genet C Semin Med Genet. 2008 Nov 15;148C(4):270-4. doi: 10.1002/ajmg.c.30188. Citation on PubMed
- South ST, Whitby H, Battaglia A, Carey JC, Brothman AR. Comprehensive analysis of Wolf-Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur J Hum Genet. 2008 Jan;16(1):45-52. doi: 10.1038/sj.ejhg.5201915. Epub 2007 Aug 29. Citation on PubMed
- Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, Butte C, Memo L, Capovilla G, Neri G. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003 Mar;72(3):590-7. doi: 10.1086/367925. Epub 2003 Jan 30. Citation on PubMed or Free article on PubMed Central
- Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008 Nov 15;148C(4):257-69. doi: 10.1002/ajmg.c.30190. Citation on PubMed
- Zollino M, Orteschi D, Ruiter M, Pfundt R, Steindl K, Cafiero C, Ricciardi S, Contaldo I, Chieffo D, Ranalli D, Acquafondata C, Murdolo M, Marangi G, Asaro A, Battaglia D. Unusual 4p16.3 deletions suggest an additional chromosome region for the Wolf-Hirschhorn syndrome-associated seizures disorder. Epilepsia. 2014 Jun;55(6):849-57. doi: 10.1111/epi.12617. Epub 2014 Apr 16. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.