

Description
Von Willebrand disease is a bleeding disorder that slows the blood clotting process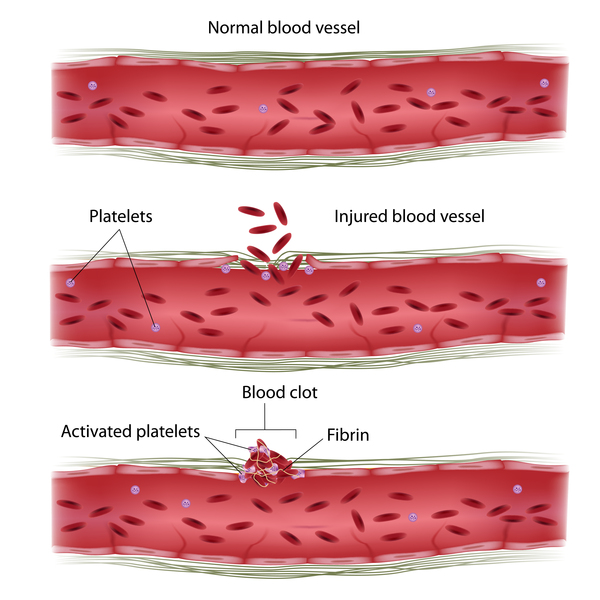 , causing prolonged bleeding after an injury. People with this condition often experience easy bruising, long-lasting nosebleeds, and excessive bleeding or oozing following an injury, surgery, or dental work. Mild forms of von Willebrand disease may become apparent only when abnormal bleeding occurs following surgery or a serious injury. People with this condition who have menstrual periods typically have heavy or prolonged bleeding during menstruation (menorrhagia), and some may also experience reproductive tract bleeding during pregnancy and childbirth. In severe cases of von Willebrand disease, heavy bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Symptoms of von Willebrand disease may change over time. Increased age, pregnancy, exercise, and stress may cause bleeding symptoms to become less frequent.
, causing prolonged bleeding after an injury. People with this condition often experience easy bruising, long-lasting nosebleeds, and excessive bleeding or oozing following an injury, surgery, or dental work. Mild forms of von Willebrand disease may become apparent only when abnormal bleeding occurs following surgery or a serious injury. People with this condition who have menstrual periods typically have heavy or prolonged bleeding during menstruation (menorrhagia), and some may also experience reproductive tract bleeding during pregnancy and childbirth. In severe cases of von Willebrand disease, heavy bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Symptoms of von Willebrand disease may change over time. Increased age, pregnancy, exercise, and stress may cause bleeding symptoms to become less frequent.
Von Willebrand disease is divided into three types. Type 1 has one subtype (1C), and type 2 is divided into four subtypes (2A, 2B, 2M, and 2N). Type 1 is the most common of the three types, accounting for 75 percent of affected individuals. Type 1 is typically mild, but some people are severely affected. Type 2 accounts for about 15 percent of cases. This type is usually of intermediate severity. Type 3 is the rarest form of the condition, accounting for about 5 percent of affected individuals, and is usually the most severe.
Another form of the disorder, acquired von Willebrand syndrome, is not caused by inherited gene variants (also called mutations). Acquired von Willebrand syndrome is typically seen in people with other disorders, such as diseases that affect bone marrow or immune cell function. This rare form of the condition is characterized by abnormal bleeding into the skin and other soft tissues, usually beginning in adulthood.
or immune cell function. This rare form of the condition is characterized by abnormal bleeding into the skin and other soft tissues, usually beginning in adulthood.
Frequency
Von Willebrand disease is estimated to affect 1 in 100 to 10,000 individuals. Because people with mild signs and symptoms are not likely to seek medical attention, this condition may be underdiagnosed. Most researchers agree that von Willebrand disease is the most common genetic bleeding disorder.
Causes
Variants in the VWF gene cause von Willebrand disease. The VWF gene provides instructions for making a blood clotting protein called von Willebrand factor, which is essential for the formation of blood clots.
After an injury, clots protect the body by sealing off damaged blood vessels and preventing further blood loss. Von Willebrand factor acts as a glue that holds blood clots together and prevents the breakdown of other blood clotting proteins. If von Willebrand factor does not function normally or too little of the protein is available, blood clots cannot form properly. Abnormally slow blood clotting causes the prolonged bleeding episodes seen in people with von Willebrand disease.
The three types of von Willebrand disease are classified based on the amount of von Willebrand factor that is produced or its ability to function. Variants in the VWF gene that reduce the amount of von Willebrand factor cause type 1 von Willebrand disease. In type 1C, the amount of von Willebrand factor is low because the protein is removed from the body more quickly than usual. People with type 1 von Willebrand disease have varying amounts of von Willebrand factor in their bloodstream. Some people with a mild case of type 1 never experience a prolonged bleeding episode.
Variants that impair the function of von Willebrand factor cause the four subtypes of type 2 von Willebrand disease. Affected individuals produce a normal amount of von Willebrand factor, but the protein cannot function properly.
In type 2A, von Willebrand factor is not the right size and cannot help form clots.
In type 2B, von Willebrand factor forms clots too easily. The clots are removed from the body, so there is not enough von Willebrand factor available when it is needed for clotting.
In types 2M and 2N, von Willebrand factor is unable to interact with other structures or proteins needed to form blood clots.
People with type 2 von Willebrand disease can have bleeding episodes of varying severity depending on the extent of the von Willebrand factor abnormalities, but the bleeding episodes are typically similar to those seen in type 1.
Variants that severely reduce or eliminate von Willebrand factor cause type 3 von Willebrand disease. People with type 3 von Willebrand disease usually have severe bleeding episodes.
Inheritance
Von Willebrand disease can have different inheritance patterns.
Most cases of type 1 and type 2 von Willebrand disease are inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Type 3, some cases of type 2, and a small number of type 1 cases of von Willebrand disease are inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Angiohemophilia
- Vascular pseudohemophilia
- Von Willebrand disorder
- Von Willebrand's factor deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Connell NT, Flood VH, Brignardello-Petersen R, Abdul-Kadir R, Arapshian A, Couper S, Grow JM, Kouides P, Laffan M, Lavin M, Leebeek FWG, O'Brien SH, Ozelo MC, Tosetto A, Weyand AC, James PD, Kalot MA, Husainat N, Mustafa RA. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021 Jan 12;5(1):301-325. doi: 10.1182/bloodadvances.2020003264. Citation on PubMed
- Federici AB, Mannucci PM. Management of inherited von Willebrand disease in 2007. Ann Med. 2007;39(5):346-58. doi: 10.1080/07853890701513738. Citation on PubMed
- James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost. 2007 Jun;5(6):1165-9. doi: 10.1111/j.1538-7836.2007.02563.x. Citation on PubMed
- James PD, Connell NT, Ameer B, Di Paola J, Eikenboom J, Giraud N, Haberichter S, Jacobs-Pratt V, Konkle B, McLintock C, McRae S, R Montgomery R, O'Donnell JS, Scappe N, Sidonio R, Flood VH, Husainat N, Kalot MA, Mustafa RA. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021 Jan 12;5(1):280-300. doi: 10.1182/bloodadvances.2020003265. Citation on PubMed
- Kessler CM. Diagnosis and treatment of von Willebrand disease: new perspectives and nuances. Haemophilia. 2007 Dec;13 Suppl 5:3-14. doi: 10.1111/j.1365-2516.2007.01581.x. No abstract available. Erratum In: Haemophilia. 2008 May;14(3):669. Citation on PubMed
- Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, Rick ME, Sadler JE, Weinstein M, Yawn BP. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008 Mar;14(2):171-232. doi: 10.1111/j.1365-2516.2007.01643.x. Citation on PubMed
- Nichols WL, Rick ME, Ortel TL, Montgomery RR, Sadler JE, Yawn BP, James AH, Hultin MB, Manco-Johnson MJ, Weinstein M. Clinical and laboratory diagnosis of von Willebrand disease: a synopsis of the 2008 NHLBI/NIH guidelines. Am J Hematol. 2009 Jun;84(6):366-70. doi: 10.1002/ajh.21405. Citation on PubMed
- Peake I, Goodeve A. Type 1 von Willebrand disease. J Thromb Haemost. 2007 Jul;5 Suppl 1:7-11. doi: 10.1111/j.1538-7836.2007.02488.x. Citation on PubMed
- Pruthi RK. A practical approach to genetic testing for von Willebrand disease. Mayo Clin Proc. 2006 May;81(5):679-91. doi: 10.4065/81.5.679. Citation on PubMed
- Weyand AC, Flood VH. Von Willebrand Disease: Current Status of Diagnosis and Management. Hematol Oncol Clin North Am. 2021 Dec;35(6):1085-1101. doi: 10.1016/j.hoc.2021.07.004. Epub 2021 Aug 13. Citation on PubMed
- Wilde JT. Von Willebrand disease. Clin Med (Lond). 2007 Dec;7(6):629-32. doi: 10.7861/clinmedicine.7-6-629. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.