

Description
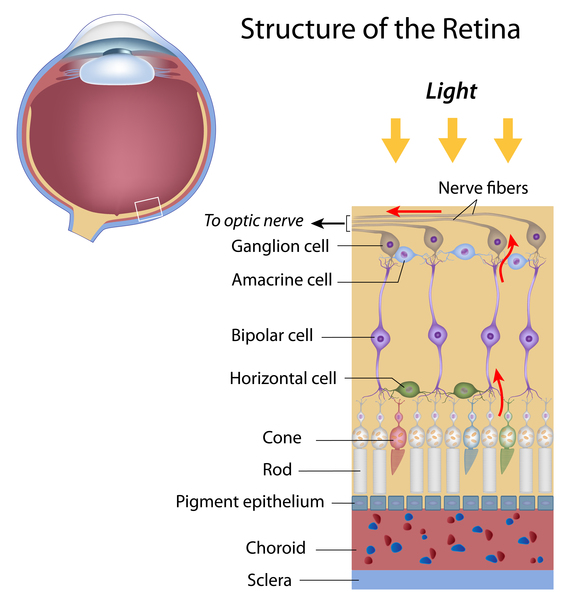
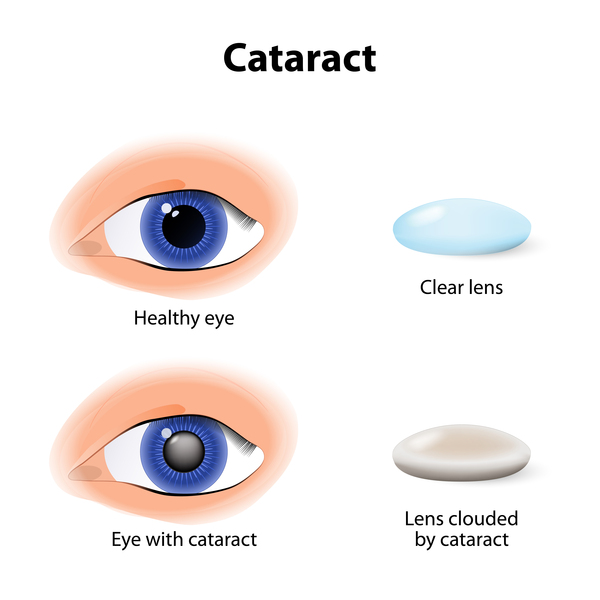
Usher syndrome is a condition characterized by partial or total hearing loss and vision loss that worsens over time. The hearing loss is classified as sensorineural, which means that it is caused by abnormalities of the inner ear. The loss of vision is caused by an eye disease called retinitis pigmentosa (RP), which affects the layer of light-sensitive tissue at the back of the eye (the retina). Vision loss occurs as the light-sensing cells of the retina gradually break down. Loss of night vision begins first, followed by blind spots that develop in the side (peripheral) vision. Over time, these blind spots enlarge and merge to produce tunnel vision. In some cases, vision is further impaired by clouding of the lens of the eye (cataracts). However, many people with retinitis pigmentosa retain some central vision throughout their lives.
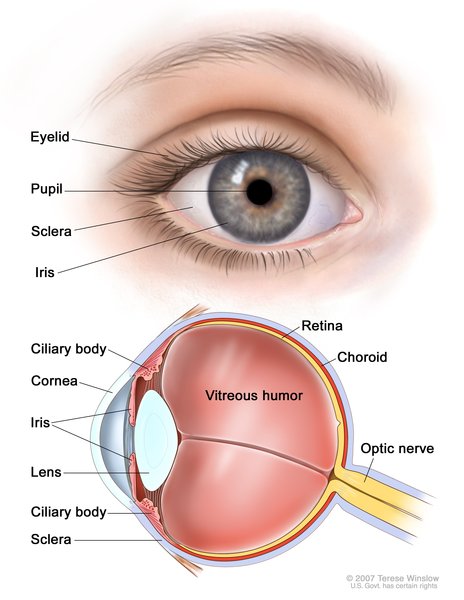
Researchers have identified three major types of Usher syndrome, designated as types I, II, and III. These types are distinguished by the severity of hearing loss, the presence or absence of balance problems, and the age at which signs and symptoms appear. The types are further divided into subtypes based on their genetic cause.
Most individuals with Usher syndrome type I are born with severe to profound hearing loss. Worsening vision loss caused by retinitis pigmentosa becomes apparent in childhood. This type of Usher syndrome also causes abnormalities of the vestibular system, which is the part of the inner ear that helps maintain the body's balance and orientation in space. As a result of the vestibular abnormalities, children with the condition have trouble with balance. They begin sitting independently and walking later than usual, and they may have difficulty riding a bicycle and playing certain sports.
Usher syndrome type II is characterized by hearing loss from birth and progressive vision loss that begins in adolescence or adulthood. The hearing loss associated with this form of Usher syndrome ranges from mild to severe and mainly affects the ability to hear high-frequency sounds. For example, it is difficult for affected individuals to hear high, soft speech sounds, such as those of the letters d and t. The degree of hearing loss varies within and among families with this condition, and it may become more severe over time. Unlike the other forms of Usher syndrome, type II is not associated with vestibular abnormalities that cause difficulties with balance.
People with Usher syndrome type III experience hearing loss and vision loss beginning somewhat later in life. Unlike the other forms of Usher syndrome, type III is usually associated with normal hearing at birth. Hearing loss typically begins during late childhood or adolescence, after the development of speech, and becomes more severe over time. By middle age, most affected individuals have profound hearing loss. Vision loss caused by retinitis pigmentosa also develops in late childhood or adolescence. Some people with Usher syndrome type III develop vestibular abnormalities that cause problems with balance.
Frequency
Usher syndrome affects around 4 to 17 in 100,000 people. Types I and II are the most common forms of Usher syndrome in most countries. Certain genetic mutations resulting in type 1 Usher syndrome are more common among people of Ashkenazi (eastern and central European) Jewish or French Acadian heritage than in the general population.
Type III represents only about 2 percent of all Usher syndrome cases overall. However, type III occurs more frequently in the Finnish population, where it accounts for about 40 percent of cases, and among people of Ashkenazi Jewish heritage.
Causes
Usher syndrome can be caused by mutations in several different genes. Mutations in at least six genes can cause Usher syndrome type I. The most common of these are MYO7A gene mutations, followed by mutations in the CDH23 gene. Usher syndrome type II can result from mutations in three genes; USH2A gene mutations account for most cases of type II. Usher syndrome type III is most often caused by mutations in the CLRN1 gene.
The genes associated with Usher syndrome provide instructions for making proteins involved in normal hearing, balance, and vision. In the inner ear, these proteins are involved in the development and function of specialized cells called hair cells, which help to transmit sound and signals from the inner ear to the brain. In the retina, the proteins contribute to the maintenance of light-sensing cells called rod photoreceptors (which provide vision in low light) and cone photoreceptors (which provide color vision and vision in bright light). For some of the proteins related to Usher syndrome, their exact role in hearing, balance, and vision is unknown.

Most of the gene mutations responsible for Usher syndrome lead to a loss of hair cells in the inner ear and a gradual loss of rods and cones in the retina. Degeneration of these sensory cells causes the hearing loss, balance problems, and vision loss that occur with Usher syndrome.
In some people with Usher syndrome, the genetic cause of the condition has not been identified. Researchers suspect that several additional genes are probably associated with this disorder.
Inheritance
All of the types of Usher syndrome are inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have a mutation. The parents of an individual with Usher syndrome each carry one copy of the mutated gene, but they do not have any signs and symptoms of the condition.

Other Names for This Condition
- Deafness-retinitis pigmentosa syndrome
- Graefe-Usher syndrome
- Hallgren syndrome
- Retinitis pigmentosa-deafness syndrome
- Usher's syndrome
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Usher syndrome

- Genetic Testing Registry: Usher syndrome type 1B
- Genetic Testing Registry: Usher syndrome type 1C
- Genetic Testing Registry: Usher syndrome type 1G
- Genetic Testing Registry: Usher syndrome type 1H
- Genetic Testing Registry: Usher syndrome type 1J
- Genetic Testing Registry: Usher syndrome type 1K
- Genetic Testing Registry: Usher syndrome type 2A
- Genetic Testing Registry: Usher syndrome type 2C
- Genetic Testing Registry: Usher syndrome type 2D
- Genetic Testing Registry: Usher syndrome type 3
- Genetic Testing Registry: Usher syndrome type 3B
- Genetic Testing Registry: Usher syndrome, type 1D/F
- Genetic Testing Registry: Usher syndrome, type IIC, GPR98/PDZD7 digenic
- Genetic Testing Registry: Usher syndrome type 1
- Genetic Testing Registry: Usher syndrome type 1D
- Genetic Testing Registry: Usher syndrome type 1E
- Genetic Testing Registry: Usher syndrome type 1F
- Genetic Testing Registry: Usher syndrome type 2
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- USHER SYNDROME, TYPE I; USH1
- USHER SYNDROME, TYPE IIA; USH2A
- USHER SYNDROME, TYPE IIIA; USH3A
- USHER SYNDROME, TYPE IC; USH1C
- USHER SYNDROME, TYPE ID; USH1D
- USHER SYNDROME, TYPE IF; USH1F
- USHER SYNDROME, TYPE IE; USH1E
- USHER SYNDROME, TYPE IIC; USH2C
- USHER SYNDROME, TYPE IG; USH1G
- DEAFNESS, AUTOSOMAL RECESSIVE 48; DFNB48
- USHER SYNDROME, TYPE IID; USH2D
- USHER SYNDROME, TYPE IH; USH1H
- USHER SYNDROME, TYPE IK; USH1K
- USHER SYNDROME, TYPE IIIB; USH3B
Scientific Articles on PubMed
References
- Aparisi MJ, Aller E, Fuster-Garcia C, Garcia-Garcia G, Rodrigo R, Vazquez-Manrique RP, Blanco-Kelly F, Ayuso C, Roux AF, Jaijo T, Millan JM. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J Rare Dis. 2014 Nov 18;9:168. doi: 10.1186/s13023-014-0168-7. Citation on PubMed or Free article on PubMed Central
- Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012 Feb;25(1):42-9. doi: 10.1097/WCO.0b013e32834ef8b2. Citation on PubMed
- Bonnet C, Grati M, Marlin S, Levilliers J, Hardelin JP, Parodi M, Niasme-Grare M, Zelenika D, Delepine M, Feldmann D, Jonard L, El-Amraoui A, Weil D, Delobel B, Vincent C, Dollfus H, Eliot MM, David A, Calais C, Vigneron J, Montaut-Verient B, Bonneau D, Dubin J, Thauvin C, Duvillard A, Francannet C, Mom T, Lacombe D, Duriez F, Drouin-Garraud V, Thuillier-Obstoy MF, Sigaudy S, Frances AM, Collignon P, Challe G, Couderc R, Lathrop M, Sahel JA, Weissenbach J, Petit C, Denoyelle F. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis. 2011 May 11;6:21. doi: 10.1186/1750-1172-6-21. Citation on PubMed or Free article on PubMed Central
- Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher syndrome: hearing loss with vision loss. Adv Otorhinolaryngol. 2011;70:56-65. doi: 10.1159/000322473. Epub 2011 Feb 24. Citation on PubMed
- Koenekoop R, Arriaga M, Trzupek KM, Lentz J. Usher Syndrome Type II. 1999 Dec 10 [updated 2023 Mar 23]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1341/ Citation on PubMed
- Koenekoop RK, Arriaga MA, Trzupek KM, Lentz JJ. Usher Syndrome Type I. 1999 Dec 10 [updated 2020 Oct 8]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1265/ Citation on PubMed
- Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2015 Mar;1852(3):406-20. doi: 10.1016/j.bbadis.2014.11.020. Epub 2014 Dec 4. Citation on PubMed or Free article on PubMed Central
- Saihan Z, Webster AR, Luxon L, Bitner-Glindzicz M. Update on Usher syndrome. Curr Opin Neurol. 2009 Feb;22(1):19-27. doi: 10.1097/wco.0b013e3283218807. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.