

Description
STXBP1 encephalopathy is a condition characterized by abnormal brain function (encephalopathy) and intellectual disability. Most affected individuals also have recurrent seizures (epilepsy). The signs and symptoms of this condition typically begin in infancy but can start later in childhood or early adulthood. In many affected individuals who have epilepsy, the seizures stop after a few years, and the other neurological problems continue throughout life. However, some people with STXBP1 encephalopathy have seizures that persist.
In people with STXBP1 encephalopathy, intellectual disability is often severe to profound. In addition, speech and motor skills, such as sitting, crawling, and walking, can be delayed. Though they may acquire the skill late, many children with the condition can walk independently by age 5. Affected individuals usually learn their first words later than their peers, sometimes not until late childhood. Some can communicate verbally using simple sentences, while others never develop the skill.
About 85 percent of people with STXBP1 encephalopathy develop epilepsy. The most common seizures in this condition are infantile spasms, which occur before age 1 and consist of involuntary muscle contractions. Other seizure types that can occur in people with this condition include uncontrolled muscle twitches (myoclonic seizures), sudden episodes of weak muscle tone (atonic seizures), partial or complete loss of consciousness (absence seizures), or loss of consciousness with muscle rigidity and convulsions (tonic-clonic seizures). Most people who have STXBP1 encephalopathy have more than one type of seizure. In about one-quarter of affected individuals, the seizures are described as refractory because they do not respond to therapy with anti-epileptic medications.
Other neurological problems that occur in people with STXBP1 encephalopathy include features of autism spectrum disorder; weak muscle tone (hypotonia); and movement problems, such as difficulty coordinating movements (ataxia), involuntary trembling (tremors), and muscle stiffness (spasticity). In some cases, areas of brain tissue loss (atrophy) have been found on medical imaging.
Frequency
The prevalence of STXBP1 encephalopathy with epilepsy is unknown. More than 280 individuals with this condition have been reported in the medical literature.
Causes
As its name indicates, STXBP1 encephalopathy is caused by mutations in the STXBP1 gene. This gene provides instructions for making syntaxin-binding protein 1. In nerve cells (neurons ), this protein helps regulate the release of chemical messengers called neurotransmitters
), this protein helps regulate the release of chemical messengers called neurotransmitters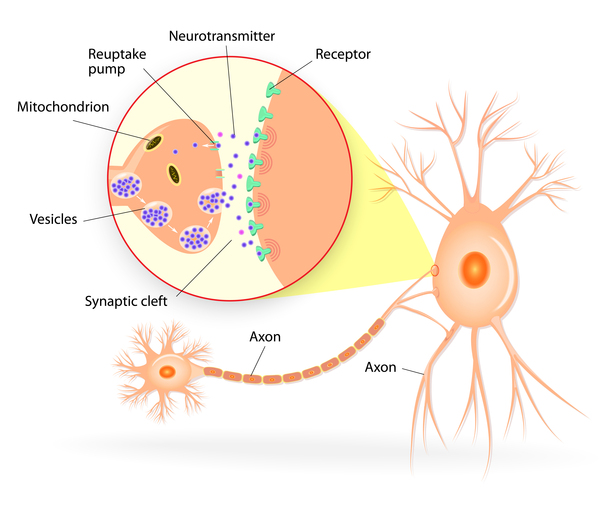 from compartments known as synaptic vesicles. The release of neurotransmitters relays signals between neurons and is critical for normal brain function. Syntaxin-binding protein 1 is also thought to play a role in the positioning and growth of neurons during brain development.
from compartments known as synaptic vesicles. The release of neurotransmitters relays signals between neurons and is critical for normal brain function. Syntaxin-binding protein 1 is also thought to play a role in the positioning and growth of neurons during brain development.
STXBP1 gene mutations reduce the amount of functional protein produced from the gene, which impairs the release of neurotransmitters. A change in neurotransmitter levels can lead to uncontrolled activation (excitation) of neurons, which causes seizures. Research suggests that a shortage of syntaxin-binding protein 1 also impairs neuron development in the brain, which could underlie encephalopathy and other neurological problems characteristic of this condition.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- DEE4
- Developmental and epileptic encephalopathy 4
- Developmental and epileptic encephalopathy, type 4
- Early-infantile epileptic encephalopathy 4
- EIEE4
- STXBP1 encephalopathy with epilepsy
- STXBP1 epileptic encephalopathy
- STXBP1-related developmental and epileptic encephalopathy
- STXBP1-related early-onset encephalopathy
- STXBP1-related epileptic encephalopathy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abramov D, Guiberson NGL, Burre J. STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies. J Neurochem. 2021 Apr;157(2):165-178. doi: 10.1111/jnc.15120. Epub 2020 Aug 4. Citation on PubMed
- Barcia G, Chemaly N, Gobin S, Milh M, Van Bogaert P, Barnerias C, Kaminska A, Dulac O, Desguerre I, Cormier V, Boddaert N, Nabbout R. Early epileptic encephalopathies associated with STXBP1 mutations: Could we better delineate the phenotype? Eur J Med Genet. 2014 Jan;57(1):15-20. doi: 10.1016/j.ejmg.2013.10.006. Epub 2013 Nov 1. Citation on PubMed
- Di Meglio C, Lesca G, Villeneuve N, Lacoste C, Abidi A, Cacciagli P, Altuzarra C, Roubertie A, Afenjar A, Renaldo-Robin F, Isidor B, Gautier A, Husson M, Cances C, Metreau J, Laroche C, Chouchane M, Ville D, Marignier S, Rougeot C, Lebrun M, de Saint Martin A, Perez A, Riquet A, Badens C, Missirian C, Philip N, Chabrol B, Villard L, Milh M. Epileptic patients with de novo STXBP1 mutations: Key clinical features based on 24 cases. Epilepsia. 2015 Dec;56(12):1931-40. doi: 10.1111/epi.13214. Epub 2015 Oct 29. Citation on PubMed
- Gupta A. STXBP1-Related EOEE - Early Onset Epilepsy AND Encephalopathy, or is it Early Onset Epileptic Encephalopathy? Epilepsy Curr. 2016 Sep-Oct;16(5):302-304. doi: 10.5698/1535-7511-16.5.302. No abstract available. Citation on PubMed or Free article on PubMed Central
- Hamada N, Iwamoto I, Tabata H, Nagata KI. MUNC18-1 gene abnormalities are involved in neurodevelopmental disorders through defective cortical architecture during brain development. Acta Neuropathol Commun. 2017 Nov 30;5(1):92. doi: 10.1186/s40478-017-0498-5. Citation on PubMed
- Patzke C, Han Y, Covy J, Yi F, Maxeiner S, Wernig M, Sudhof TC. Analysis of conditional heterozygous STXBP1 mutations in human neurons. J Clin Invest. 2015 Sep;125(9):3560-71. doi: 10.1172/JCI78612. Epub 2015 Aug 17. Citation on PubMed or Free article on PubMed Central
- Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, Benkel-Herrenbrueck I, Benoit V, Budetta M, Caliebe A, Cantalupo G, Capovilla G, Casara G, Courage C, Deprez M, Destree A, Dilena R, Erasmus CE, Fannemel M, Fjaer R, Giordano L, Helbig KL, Heyne HO, Klepper J, Kluger GJ, Lederer D, Lodi M, Maier O, Merkenschlager A, Michelberger N, Minetti C, Muhle H, Phalin J, Ramsey K, Romeo A, Schallner J, Schanze I, Shinawi M, Sleegers K, Sterbova K, Syrbe S, Traverso M, Tzschach A, Uldall P, Van Coster R, Verhelst H, Viri M, Winter S, Wolff M, Zenker M, Zoccante L, De Jonghe P, Helbig I, Striano P, Lemke JR, Moller RS, Weckhuysen S. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology. 2016 Mar 8;86(10):954-62. doi: 10.1212/WNL.0000000000002457. Epub 2016 Feb 10. Citation on PubMed
- Yamamoto T, Shimojima K, Yano T, Ueda Y, Takayama R, Ikeda H, Imai K. Loss-of-function mutations of STXBP1 in patients with epileptic encephalopathy. Brain Dev. 2016 Mar;38(3):280-4. doi: 10.1016/j.braindev.2015.09.004. Epub 2015 Sep 16. Citation on PubMed
- Yamashita S, Chiyonobu T, Yoshida M, Maeda H, Zuiki M, Kidowaki S, Isoda K, Morimoto M, Kato M, Saitsu H, Matsumoto N, Nakahata T, Saito MK, Hosoi H. Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia. 2016 Apr;57(4):e81-6. doi: 10.1111/epi.13338. Epub 2016 Feb 25. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.