

Description
SRD5A3-congenital disorder of glycosylation (SRD5A3-CDG, formerly known as congenital disorder of glycosylation type Iq) is an inherited condition that causes neurological and vision problems and other signs and symptoms. The pattern and severity of this condition's features vary widely among affected individuals.
Individuals with SRD5A3-CDG typically develop signs and symptoms of the condition during infancy or early childhood. Most individuals with SRD5A3-CDG have intellectual disability, vision problems, unusual facial features, low muscle tone (hypotonia), and problems with coordination and balance (ataxia).
Vision problems in SRD5A3-CDG often include involuntary side-side movements of the eyes
(nystagmus), a gap or hole in one of the structures of the eye (coloboma ), underdevelopment of the nerves that carry signals between the eyes and the brain
), underdevelopment of the nerves that carry signals between the eyes and the brain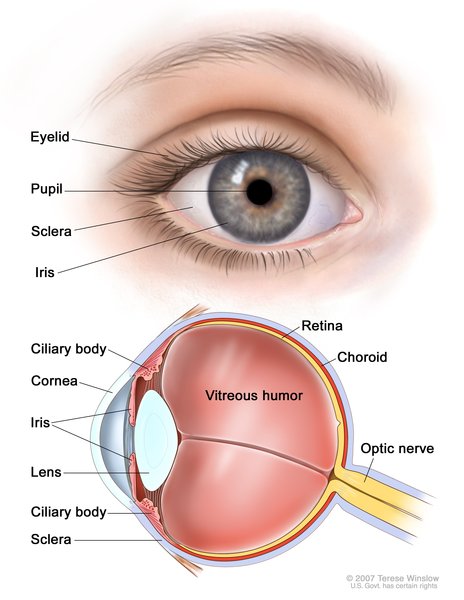 (optic nerve hypoplasia), or vision loss early in life (early-onset severe retinal dystrophy). Over time, affected individuals may develop clouding of the lenses of the eyes (cataracts
(optic nerve hypoplasia), or vision loss early in life (early-onset severe retinal dystrophy). Over time, affected individuals may develop clouding of the lenses of the eyes (cataracts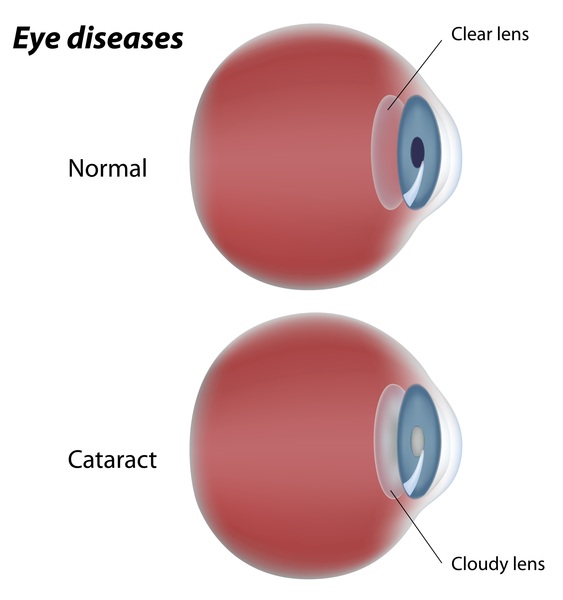 ) or increased pressure in the eyes (glaucoma
) or increased pressure in the eyes (glaucoma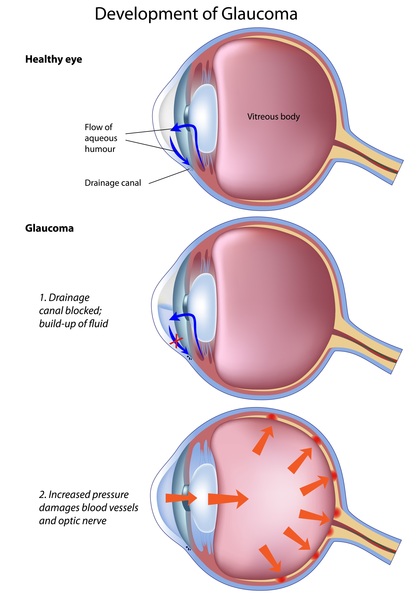 ).
).
Other features of SRD5A3-CDG can include skin rash, unusually small red blood cells (microcytic anemia),
and liver problems.
problems.
Frequency
Causes
SRD5A3-CDG is caused by variants (also known as mutations) in the SRD5A3 gene. This gene provides instructions for making the enzyme steroid 5 alpha-reductase 3, which facilitates the conversion of a compound called polyprenol to a compound called dolichol. This conversion is critical for a process called glycosylation, by which groups of sugar molecules (oligosaccharides) are attached to proteins. Glycosylation changes proteins in ways that are important for their functions.
Variants in the SRD5A3 gene typically lead to the production of abnormally small steroid 5 alpha-reductase 3 enzyme that either has no activity or is quickly broken down. Without any normal steroid 5 alpha-reductase 3 enzyme, dolichol production is impaired, and glycosylation cannot proceed normally. The signs and symptoms of SRD5A3-CDG are likely due to impaired glycosylation of proteins that are needed for the normal function of various organs and tissues.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- CDG Iq
- CDG-Iq
- Congenital disorder of glycosylation type 1q
- SRD5A3-CDG
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ben Ayed I, Ouarda W, Frikha F, Kammoun F, Souissi A, Ben Said M, Bouzid A, Elloumi I, Hamdani TM, Gharbi N, Baklouti N, Guirat M, Mejdoub F, Kharrat N, Boujelbene I, Abdelhedi F, Belguith N, Keskes L, Gibriel AA, Kamoun H, Triki C, Alimi AM, Masmoudi S. SRD5A3-CDG: 3D structure modeling, clinical spectrum, and computer-based dysmorphic facial recognition. Am J Med Genet A. 2021 Apr;185(4):1081-1090. doi: 10.1002/ajmg.a.62065. Epub 2021 Jan 6. Citation on PubMed
- Jaeken J, Lefeber DJ, Matthijs G. SRD5A3 defective congenital disorder of glycosylation: clinical utility gene card. Eur J Hum Genet. 2020 Sep;28(9):1297-1300. doi: 10.1038/s41431-020-0647-3. Epub 2020 May 18. No abstract available. Citation on PubMed
- Kara B, Ayhan O, Gokcay G, Basbogaoglu N, Tolun A. Adult phenotype and further phenotypic variability in SRD5A3-CDG. BMC Med Genet. 2014 Jan 16;15:10. doi: 10.1186/1471-2350-15-10. Citation on PubMed
- Kousal B, Honzik T, Hansikova H, Ondruskova N, Cechova A, Tesarova M, Stranecky V, Meliska M, Michaelides M, Liskova P. Review of SRD5A3 Disease-Causing Sequence Variants and Ocular Findings in Steroid 5alpha-Reductase Type 3 Congenital Disorder of Glycosylation, and a Detailed New Case. Folia Biol (Praha). 2019;65(3):134-141. doi: 10.14712/fb2019065030134. Citation on PubMed
- Lipinski P, Cielecka-Kuszyk J, Czarnowska E, Bogdanska A, Socha P, Tylki-Szymanska A. Congenital disorders of glycosylation in children - Histopathological and ultrastructural changes in the liver. Pediatr Neonatol. 2021 May;62(3):278-283. doi: 10.1016/j.pedneo.2021.01.017. Epub 2021 Feb 5. Citation on PubMed
- Morava E, Wevers RA, Cantagrel V, Hoefsloot LH, Al-Gazali L, Schoots J, van Rooij A, Huijben K, van Ravenswaaij-Arts CM, Jongmans MC, Sykut-Cegielska J, Hoffmann GF, Bluemel P, Adamowicz M, van Reeuwijk J, Ng BG, Bergman JE, van Bokhoven H, Korner C, Babovic-Vuksanovic D, Willemsen MA, Gleeson JG, Lehle L, de Brouwer AP, Lefeber DJ. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010 Nov;133(11):3210-20. doi: 10.1093/brain/awq261. Epub 2010 Sep 17. Citation on PubMed
- Taylor RL, Arno G, Poulter JA, Khan KN, Morarji J, Hull S, Pontikos N, Rueda Martin A, Smith KR, Ali M, Toomes C, McKibbin M, Clayton-Smith J, Grunewald S, Michaelides M, Moore AT, Hardcastle AJ, Inglehearn CF, Webster AR, Black GC; UK Inherited Retinal Disease Consortium and the 100,000 Genomes Project. Association of Steroid 5alpha-Reductase Type 3 Congenital Disorder of Glycosylation With Early-Onset Retinal Dystrophy. JAMA Ophthalmol. 2017 Apr 1;135(4):339-347. doi: 10.1001/jamaophthalmol.2017.0046. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.