

Description
Spinocerebellar ataxia type 6 (SCA6) is a condition characterized by progressive problems with movement. People with this condition initially experience problems with coordination and balance (ataxia). Other early signs and symptoms of SCA6 include speech difficulties, involuntary eye movements (nystagmus), and double vision. Over time, individuals with SCA6 may develop loss of coordination in their arms, tremors, and uncontrolled muscle tensing (dystonia).
Signs and symptoms of SCA6 typically begin in a person's forties or fifties but can appear anytime from childhood to late adulthood. People with this disorder may require walking or mobility assistance later in life.
Frequency
The worldwide prevalence of SCA6 is estimated to be less than 1 in 100,000 individuals.
Causes
Variants (also known as mutations) in the CACNA1A gene cause SCA6. The CACNA1A gene provides instructions for making a protein that forms a part of some calcium channels. These channels transport positively charged calcium atoms (calcium ions) across cell membranes. The movement of these ions is critical for normal signaling between nerve cells (neurons) in the brain and other parts of the nervous system. The CACNA1A gene provides instructions for making one part (the alpha-1 subunit) of a calcium channel called CaV2.1. CaV2.1 channels play an essential role in communication between neurons in the brain.


The CACNA1A gene variants that cause SCA6 involve a DNA segment known as a CAG trinucleotide repeat. This segment is made up of a series of three DNA building blocks (cytosine, adenine, and guanine) that appear multiple times in a row. Normally, the CAG segment is repeated 4 to 18 times within the gene. In people with SCA6, the CAG segment is repeated 20 to 33 times. People with 20 repeats tend to experience signs and symptoms of SCA6 beginning in late adulthood, while people with a larger number of repeats usually have signs and symptoms from mid-adulthood.
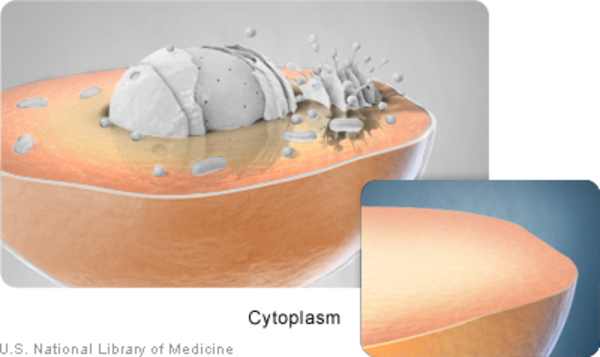
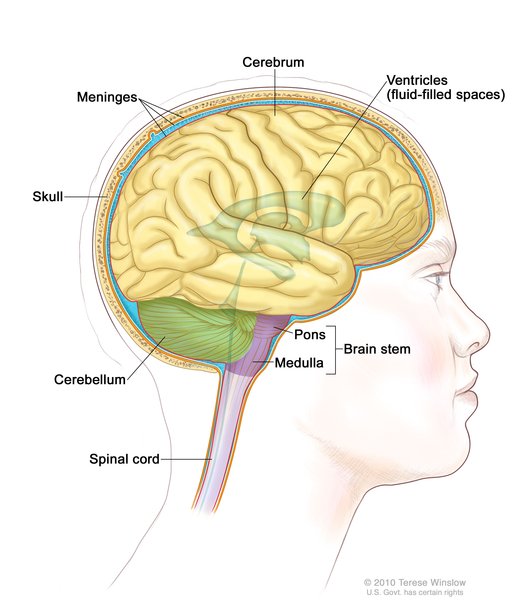
An increase in the length of the CAG segment leads to the production of an abnormally long version of the alpha-1 subunit. This version of the subunit alters the location and function of the CaV2.1 channels. Normally the alpha-1 subunit is located within the cell membrane; the abnormal subunit is found in the cell membrane as well as in the fluid inside cells (cytoplasm), where it clusters together and forms clumps (aggregates). The effect these aggregates have on cell functioning is unknown. The lack of normal calcium channels in the cell membrane impairs cell communication between neurons in the brain. Diminished cell communication leads to cell death. Cells within the cerebellum, which is the part of the brain that coordinates movement, are particularly sensitive to the accumulation of these aggregates. Over time, a loss of cells in the cerebellum causes the movement problems characteristic of SCA6.
Inheritance
This condition is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
As the altered CACNA1A gene is passed down from one generation to the next, the length of the CAG trinucleotide repeat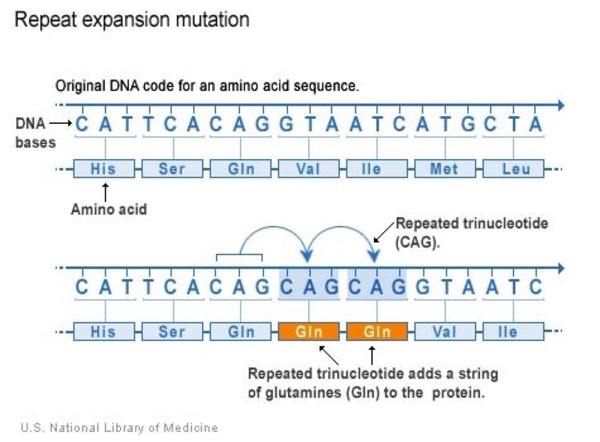 often slightly increases. A larger number of repeats is usually associated with an earlier onset of signs and symptoms. This phenomenon is called anticipation.
often slightly increases. A larger number of repeats is usually associated with an earlier onset of signs and symptoms. This phenomenon is called anticipation.
Other Names for This Condition
- SCA6
- Type 6 spinocerebellar ataxia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Casey HL, Gomez CM. Spinocerebellar Ataxia Type 6. 1998 Oct 23 [updated 2019 Nov 21]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1140/ Citation on PubMed
- Ishiguro T, Ishikawa K, Takahashi M, Obayashi M, Amino T, Sato N, Sakamoto M, Fujigasaki H, Tsuruta F, Dolmetsch R, Arai T, Sasaki H, Nagashima K, Kato T, Yamada M, Takahashi H, Hashizume Y, Mizusawa H. The carboxy-terminal fragment of alpha(1A) calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol. 2010 Apr;119(4):447-64. doi: 10.1007/s00401-009-0630-0. Epub 2009 Dec 31. Citation on PubMed or Free article on PubMed Central
- Kordasiewicz HB, Gomez CM. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics. 2007 Apr;4(2):285-94. doi: 10.1016/j.nurt.2007.01.003. Citation on PubMed
- Rajakulendran S, Schorge S, Kullmann DM, Hanna MG. Dysfunction of the Ca(V)2.1 calcium channel in cerebellar ataxias. F1000 Biol Rep. 2010 Jan 18;2:4. doi: 10.3410/B2-4. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.