

Description
Spinal muscular atrophy with lower extremity predominance (SMA-LED) is characterized by muscle weakness and wasting (atrophy) in the lower limbs, most severely affecting the thigh muscles (quadriceps). (In SMA-LED, the "D" stands for dominant, which refers to the inheritance pattern of this condition.) The loss of nerve cells that control muscle movement (motor neurons) leads to atrophy of the muscles in the lower limbs. Affected individuals often have a waddling or unsteady walk and walk on the balls of their feet. They may have difficulty rising from a seated position and climbing stairs. Some people with SMA-LED also have weakness in upper limb muscles.
Joint deformities (contractures) in the hips, knees, feet, and ankles can occur in SMA-LED, and in severe cases are present from birth and can impair walking. Some individuals with this disorder have rigidity of joints (arthrogryposis) in their shoulders, elbows, and hands.
In most people with SMA-LED, the muscle problems are apparent in infancy or early childhood; however, about one-quarter of affected individuals do not develop muscle weakness until adulthood. The muscle weakness and related health problems typically do not worsen over time.
Frequency
SMA-LED is thought to be a rare condition; its prevalence is unknown.
Causes
SMA-LED is caused by mutations in the DYNC1H1 gene or BICD2 gene. When this condition is caused by mutations in the DYNC1H1 gene, it is often known as SMA-LED type 1 or SMA-LED1, and when it is caused by mutations in the BICD2 gene, the condition is often known as SMA-LED type 2 or SMA-LED2.
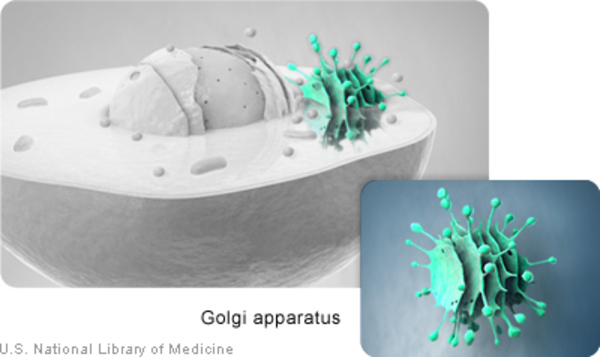
The DYNC1H1 gene provides instructions for making a protein that is part of a group (complex) of proteins called dynein. This complex is part of a network that moves (transports) proteins and other materials within cells. The protein produced from the BICD2 gene attaches (binds) to the dynein complex, turning it on (activating it) and helping it bind to other cellular materials for transport. The BICD2 protein and the dynein complex help neighboring neurons communicate by transporting sac-like structures called synaptic vesicles that contain chemical messengers. The BICD2 protein also helps maintain the structure of a cell component called the Golgi apparatus, in which newly produced proteins are modified so they can carry out their functions.
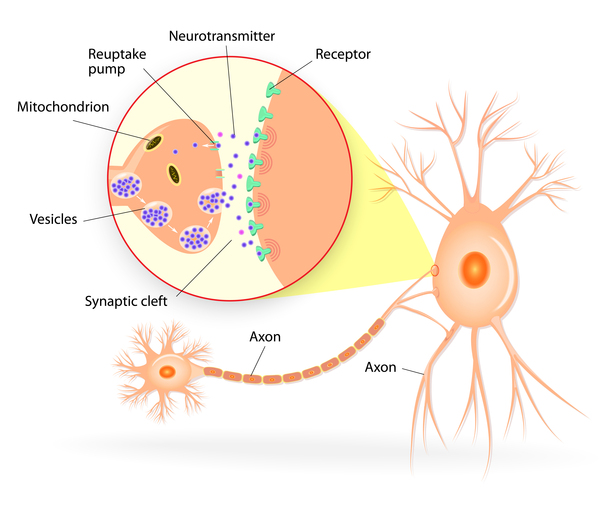
DYNC1H1 and BICD2 gene mutations that cause SMA-LED disrupt the function of the dynein complex. As a result, transport of proteins, synaptic vesicles, and other materials within cells is reduced. Decreased synaptic vesicle transport in motor neurons, leading to impaired growth of neurons, is thought to contribute to the muscle weakness and atrophy experienced by people with SMA-LED. It is unclear why this condition primarily affects the lower limbs.
Additionally, BICD2 gene mutations impair the BICD2 protein's ability to maintain the structure of the Golgi apparatus within cells. As a result, the Golgi apparatus breaks down into small fragments and the altered BICD2 protein becomes trapped within these fragments. Loss of these cell components likely further contributes to the signs and symptoms of SMA-LED.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Other Names for This Condition
- Autosomal dominant childhood-onset proximal spinal muscular atrophy with contractures
- Kugelberg-Welander syndrome, autosomal dominant
- Lower extremity-predominant autosomal dominant proximal spinal muscular atrophy with contractures
- SMA-LED
- Spinal muscular atrophy, childhood, proximal, autosomal dominant
- Spinal muscular atrophy, juvenile, proximal, autosomal dominant
- Spinal muscular atrophy, lower extremity, autosomal dominant
- Spinal muscular atrophy, lower extremity, dominant
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM, Miller LJ, Jani-Acsadi A, Pestronk A, Shy ME, Muntoni F, Vallee RB, Baloh RH. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012 May 29;78(22):1714-20. doi: 10.1212/WNL.0b013e3182556c05. Epub 2012 Mar 28. Citation on PubMed or Free article on PubMed Central
- Martinez-Carrera LA, Wirth B. Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein. Front Neurosci. 2015 Nov 5;9:401. doi: 10.3389/fnins.2015.00401. eCollection 2015. Citation on PubMed or Free article on PubMed Central
- Neveling K, Martinez-Carrera LA, Holker I, Heister A, Verrips A, Hosseini-Barkooie SM, Gilissen C, Vermeer S, Pennings M, Meijer R, te Riele M, Frijns CJ, Suchowersky O, MacLaren L, Rudnik-Schoneborn S, Sinke RJ, Zerres K, Lowry RB, Lemmink HH, Garbes L, Veltman JA, Schelhaas HJ, Scheffer H, Wirth B. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet. 2013 Jun 6;92(6):946-54. doi: 10.1016/j.ajhg.2013.04.011. Epub 2013 May 9. Citation on PubMed or Free article on PubMed Central
- Rossor AM, Oates EC, Salter HK, Liu Y, Murphy SM, Schule R, Gonzalez MA, Scoto M, Phadke R, Sewry CA, Houlden H, Jordanova A, Tournev I, Chamova T, Litvinenko I, Zuchner S, Herrmann DN, Blake J, Sowden JE, Acsadi G, Rodriguez ML, Menezes MP, Clarke NF, Auer Grumbach M, Bullock SL, Muntoni F, Reilly MM, North KN. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain. 2015 Feb;138(Pt 2):293-310. doi: 10.1093/brain/awu356. Epub 2014 Dec 14. Citation on PubMed or Free article on PubMed Central
- Scoto M, Rossor AM, Harms MB, Cirak S, Calissano M, Robb S, Manzur AY, Martinez Arroyo A, Rodriguez Sanz A, Mansour S, Fallon P, Hadjikoumi I, Klein A, Yang M, De Visser M, Overweg-Plandsoen WC, Baas F, Taylor JP, Benatar M, Connolly AM, Al-Lozi MT, Nixon J, de Goede CG, Foley AR, Mcwilliam C, Pitt M, Sewry C, Phadke R, Hafezparast M, Chong WK, Mercuri E, Baloh RH, Reilly MM, Muntoni F. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. 2015 Feb 17;84(7):668-79. doi: 10.1212/WNL.0000000000001269. Epub 2015 Jan 21. Citation on PubMed or Free article on PubMed Central
- Storbeck M, Horsberg Eriksen B, Unger A, Holker I, Aukrust I, Martinez-Carrera LA, Linke WA, Ferbert A, Heller R, Vorgerd M, Houge G, Wirth B. Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet. 2017 Sep;25(9):1040-1048. doi: 10.1038/ejhg.2017.98. Epub 2017 Jun 21. Citation on PubMed or Free article on PubMed Central
- Strickland AV, Schabhuttl M, Offenbacher H, Synofzik M, Hauser NS, Brunner-Krainz M, Gruber-Sedlmayr U, Moore SA, Windhager R, Bender B, Harms M, Klebe S, Young P, Kennerson M, Garcia AS, Gonzalez MA, Zuchner S, Schule R, Shy ME, Auer-Grumbach M. Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J Neurol. 2015 Sep;262(9):2124-34. doi: 10.1007/s00415-015-7727-2. Epub 2015 Jun 24. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.