

Description
Smith-Lemli-Opitz syndrome is a developmental disorder that affects many parts of the body. This condition is characterized by distinctive facial features, small head size (microcephaly), intellectual disability or learning problems, and behavioral problems. Many affected children have the characteristic features of autism, a developmental condition that affects communication and social interaction. Malformations of the heart, lungs, kidneys, gastrointestinal tract, and genitalia are also common. Infants with Smith-Lemli-Opitz syndrome have weak muscle tone (hypotonia), experience feeding difficulties, and tend to grow more slowly than other infants. Most affected individuals have fused second and third toes (syndactyly), and some have extra fingers or toes (polydactyly).

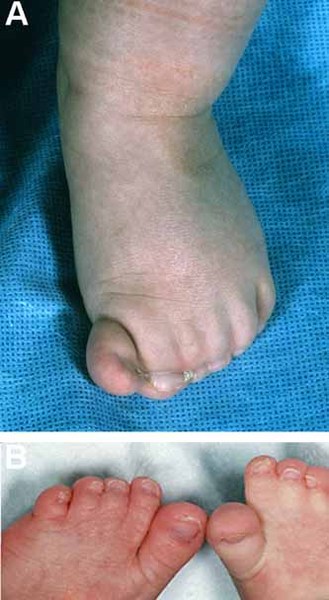
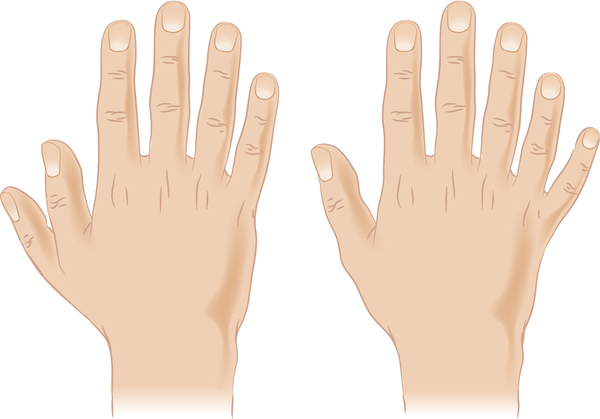
The signs and symptoms of Smith-Lemli-Opitz syndrome vary widely. Mildly affected individuals may have only minor physical abnormalities with learning and behavioral problems. Severe cases can be life-threatening and involve profound intellectual disability and major physical abnormalities.
Frequency
Smith-Lemli-Opitz syndrome affects an estimated 1 in 20,000 to 60,000 newborns. This condition is most common in whites of European ancestry, particularly people from Central European countries such as Slovakia and the Czech Republic. It is very rare among African and Asian populations.
Causes
Smith-Lemli-Opitz syndrome is caused by mutations in the DHCR7 gene, which provides instructions for making an enzyme called 7-dehydrocholesterol reductase. This enzyme is responsible for the final step in the production of cholesterol. Cholesterol is a waxy, fat-like substance that is produced in the body and obtained from foods that come from animals (particularly egg yolks, meat, poultry, fish, and dairy products). Cholesterol is necessary for normal embryonic development and has important functions both before and after birth. It is a structural component of cell membranes and the protective substance covering nerve cells (myelin). Additionally, cholesterol plays a role in the production of certain hormones and digestive acids.
Mutations in the DHCR7 gene reduce or eliminate the activity of 7-dehydrocholesterol reductase, preventing cells from producing enough cholesterol. A lack of this enzyme also allows toxic byproducts of cholesterol production to build up in the blood, nervous system, and other tissues. The combination of low cholesterol levels and an accumulation of other substances likely disrupts the growth and development of many body systems. It is not completely understood, however, how either abnormality leads to the specific features of Smith-Lemli-Opitz syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- 7-dehydrocholesterol reductase deficiency
- RSH Syndrome
- SLO syndrome
- SLOS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bianconi SE, Cross JL, Wassif CA, Porter FD. Pathogenesis, Epidemiology, Diagnosis and Clinical Aspects of Smith-Lemli-Opitz Syndrome. Expert Opin Orphan Drugs. 2015 Mar;3(3):267-280. doi: 10.1517/21678707.2015.1014472. Citation on PubMed or Free article on PubMed Central
- Jira PE, Waterham HR, Wanders RJ, Smeitink JA, Sengers RC, Wevers RA. Smith-Lemli-Opitz syndrome and the DHCR7 gene. Ann Hum Genet. 2003 May;67(Pt 3):269-80. doi: 10.1046/j.1469-1809.2003.00034.x. Citation on PubMed
- Nowaczyk MJ, Waye JS, Douketis JD. DHCR7 mutation carrier rates and prevalence of the RSH/Smith-Lemli-Opitz syndrome: where are the patients? Am J Med Genet A. 2006 Oct 1;140(19):2057-62. doi: 10.1002/ajmg.a.31413. Citation on PubMed
- Nowaczyk MJ, Waye JS. The Smith-Lemli-Opitz syndrome: a novel metabolic way of understanding developmental biology, embryogenesis, and dysmorphology. Clin Genet. 2001 Jun;59(6):375-86. doi: 10.1034/j.1399-0004.2001.590601.x. Citation on PubMed
- Nowaczyk MJM, Wassif CA. Smith-Lemli-Opitz Syndrome. 1998 Nov 13 [updated 2020 Jan 30]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1143/ Citation on PubMed
- Porter FD. RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol Genet Metab. 2000 Sep-Oct;71(1-2):163-74. doi: 10.1006/mgme.2000.3069. Citation on PubMed
- Sikora DM, Pettit-Kekel K, Penfield J, Merkens LS, Steiner RD. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2006 Jul 15;140(14):1511-8. doi: 10.1002/ajmg.a.31294. Citation on PubMed
- Waterham HR, Hennekam RC. Mutational spectrum of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 2012 Nov 15;160C(4):263-84. doi: 10.1002/ajmg.c.31346. Epub 2012 Oct 5. Citation on PubMed
- Yu H, Patel SB. Recent insights into the Smith-Lemli-Opitz syndrome. Clin Genet. 2005 Nov;68(5):383-91. doi: 10.1111/j.1399-0004.2005.00515.x. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.