

Description
SLC35A2-congenital disorder of glycosylation (SLC35A2-CDG, formerly known as congenital disorder of glycosylation type IIm) is an inherited condition that causes neurological problems and other abnormalities. This disorder's signs and symptoms and their severity vary among affected individuals.
Individuals with SLC35A2-CDG typically develop signs and symptoms of the condition early in infancy. Seizures develop within the first months of life, usually involving uncontrollable muscle stiffening (infantile spasms) that can switch to shorter episodes of muscle jerks (epileptic spasms) later in childhood. In some individuals, the seizures do not improve with anti-epileptic medications. Individuals with SLC35A2-CDG often have abnormal brain function (encephalopathy), unusual facial features, skeletal abnormalities, and weak muscle tone (hypotonia) with poor head control. They also have severe intellectual disability and delayed development, often only being able to sit or crawl and never developing meaningful speech. Affected children may have feeding difficulties and fail to grow or gain weight at the expected rate. Some have vision or hearing problems.
In SLC35A2-CDG, medical imaging shows loss of tissue (atrophy) in parts of the brain called the cerebrum and cerebellum. These brain regions are necessary for thinking ability, hearing, vision, emotion, and coordinated movement. There can also be thinning of the tissue that connects the left and right halves of the brain (the corpus callosum) or a fluid-filled sac (cyst) on the membrane that surrounds the brain (arachnoid pouch).
Frequency
SLC35A2-CDG is a rare disorder. At least nine affected individuals have been described.
Causes
Mutations in the SLC35A2 gene cause SLC35A2-CDG. This gene provides instructions for making an enzyme that is involved in a process called glycosylation. During this process, complex chains of sugar molecules (oligosaccharides) are added to proteins and fats (lipids). Glycosylation modifies proteins and lipids so they can fully perform their functions. The enzyme produced from the SLC35A2 gene transfers a simple sugar called galactose to growing oligosaccharides at a particular step in the formation of the sugar chain. Once the correct number of sugar molecules are linked together, the oligosaccharide is attached to a protein or lipid.
SLC35A2 gene mutations lead to the production of an abnormal enzyme with reduced or no activity. Without a properly functioning enzyme, glycosylation cannot proceed normally, and oligosaccharides are incomplete. The signs and symptoms of SLC35A2-CDG are likely due to impaired glycosylation of proteins and fats that are needed for the normal function of various organs and tissues.
In some individuals with SLC35A2-CDG, glycosylation becomes normal later in childhood. The cause of this apparent correction is unknown. The restoration of glycosylation in these individuals, however, does not seem to improve the signs and symptoms of SLC35A2-CDG.
Inheritance
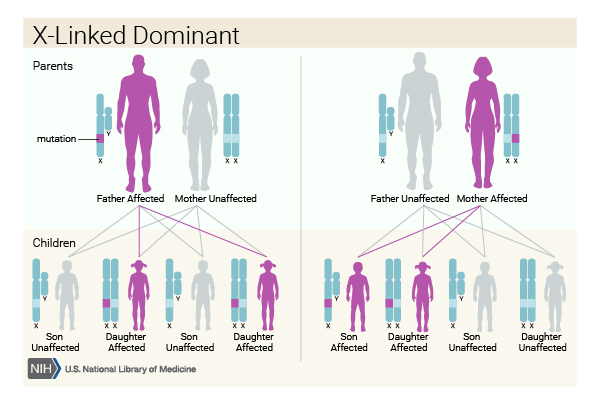
SLC35A2-CDG is inherited in an X-linked dominant pattern. The SLC35A2 gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell is thought to be incompatible with life. A few males with SLC35A2-CDG have a mutation in only some of the body's cells, a situation known as mosaicism. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

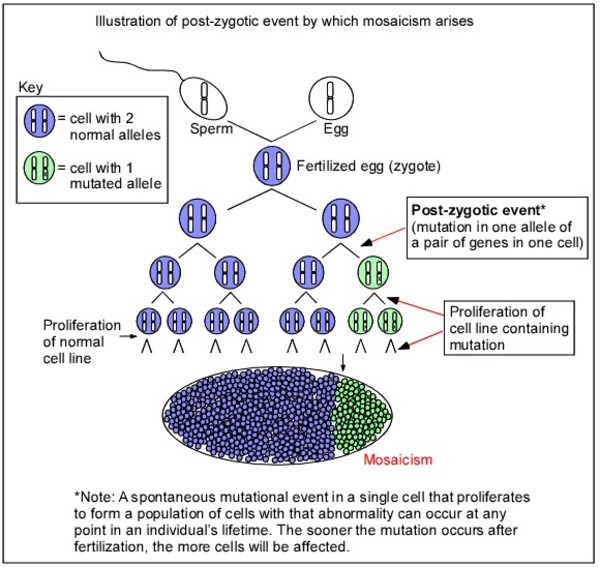
Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, so that each X chromosome is active in about half the body's cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
In SLC35A2-CDG, skewed X-inactivation allows for the normal copy of the gene to be active (expressed) and leads to the production of the normal enzyme in most cells in affected females. However, it is thought that X inactivation in nerve cells in the brain might not be skewed and so the mutated SLC35A2 gene is expressed in these cells. As a result, little or no functional enzyme is present in nerve cells, leading to the various neurological features of SLC35A2-CDG.
Other Names for This Condition
- CDG IIm
- CDG syndrome type IIm
- CDG-IIm
- CDG2M
- CDGIIm
- Congenital disorder of glycosylation, type IIm
- EIEE22
- Epileptic encephalopathy, early infantile, 22
- SLC35A2-CDG
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dorre K, Olczak M, Wada Y, Sosicka P, Gruneberg M, Reunert J, Kurlemann G, Fiedler B, Biskup S, Hortnagel K, Rust S, Marquardt T. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015 Sep;38(5):931-40. doi: 10.1007/s10545-015-9828-6. Epub 2015 Mar 17. Citation on PubMed
- Kimizu T, Takahashi Y, Oboshi T, Horino A, Koike T, Yoshitomi S, Mori T, Yamaguchi T, Ikeda H, Okamoto N, Nakashima M, Saitsu H, Kato M, Matsumoto N, Imai K. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev. 2017 Mar;39(3):256-260. doi: 10.1016/j.braindev.2016.09.009. Epub 2016 Oct 12. Citation on PubMed
- Kodera H, Nakamura K, Osaka H, Maegaki Y, Haginoya K, Mizumoto S, Kato M, Okamoto N, Iai M, Kondo Y, Nishiyama K, Tsurusaki Y, Nakashima M, Miyake N, Hayasaka K, Sugahara K, Yuasa I, Wada Y, Matsumoto N, Saitsu H. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum Mutat. 2013 Dec;34(12):1708-14. doi: 10.1002/humu.22446. Epub 2013 Oct 15. Citation on PubMed
- Ng BG, Buckingham KJ, Raymond K, Kircher M, Turner EH, He M, Smith JD, Eroshkin A, Szybowska M, Losfeld ME, Chong JX, Kozenko M, Li C, Patterson MC, Gilbert RD, Nickerson DA, Shendure J, Bamshad MJ; University of Washington Center for Mendelian Genomics; Freeze HH. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. 2013 Apr 4;92(4):632-6. doi: 10.1016/j.ajhg.2013.03.012. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.