

Description
Saul-Wilson syndrome is characterized by short stature (dwarfism) and other skeletal abnormalities. The growth problems in Saul-Wilson syndrome are called primordial, which means they begin before birth; affected individuals show slow prenatal growth (intrauterine growth retardation). After birth, affected individuals continue to grow at a very slow rate, with the average adult height being 3 feet, 6 inches (107 centimeters).
Individuals with Saul-Wilson syndrome have distinctive facial features that often include a prominent forehead, sparse scalp hair and eyebrows, prominent scalp veins, a narrow nasal bridge, a beaked nose, a wide area separating the nostrils (broad columella), a thin upper lip, and a small lower jaw (micrognathia). This combination of facial features can give affected individuals an appearance of premature aging, particularly in infancy, that is sometimes described as progeroid.
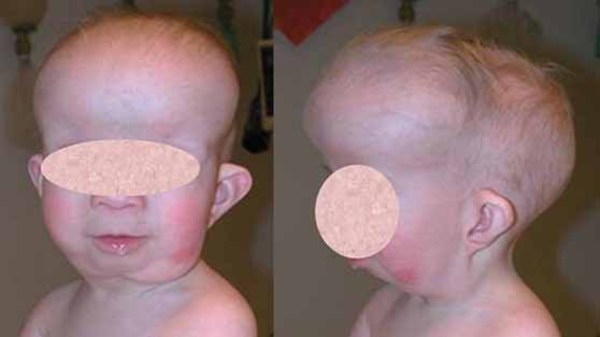

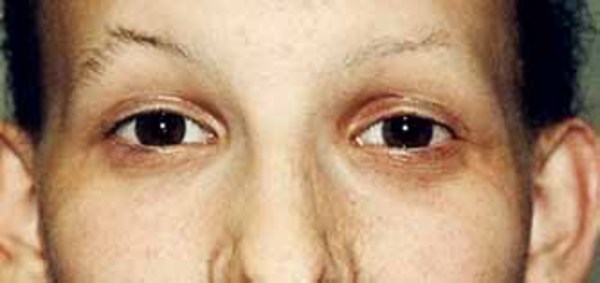
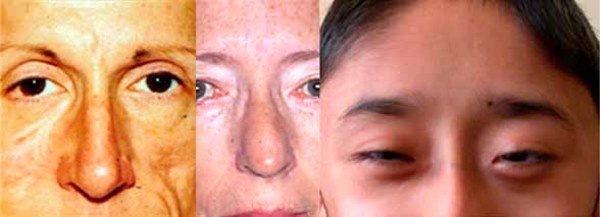


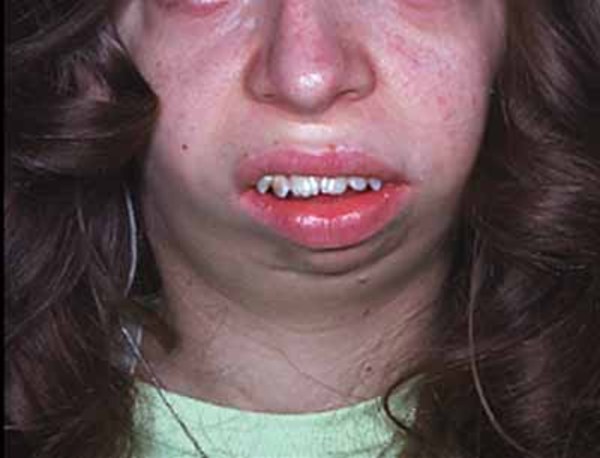
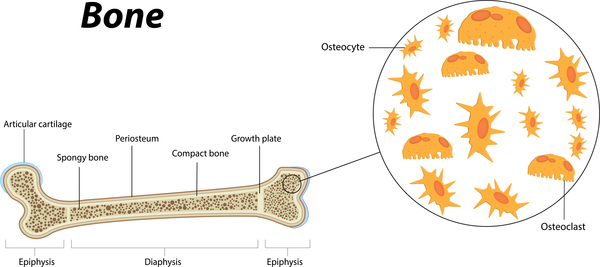
Additional skeletal abnormalities in Saul-Wilson syndrome include abnormalities in the structure of the long bones, short fingers and toes, an inward- and downward-turning foot (clubfoot), an abnormality of the hip joint that causes a decreased angle between the head and shaft of the upper leg bones (coxa vara), or flattened bones of the spine (platyspondyly) and other spinal abnormalities. Some affected individuals have bones that are unusually fragile, resulting in bone fractures that occur with little or no trauma. Adults with Saul-Wilson syndrome may experience joint pain (osteoarthritis) due to breakdown (degeneration) of the joints.

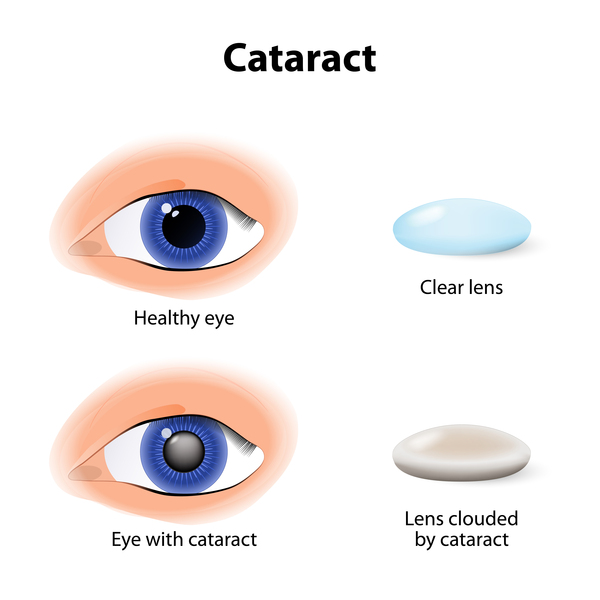
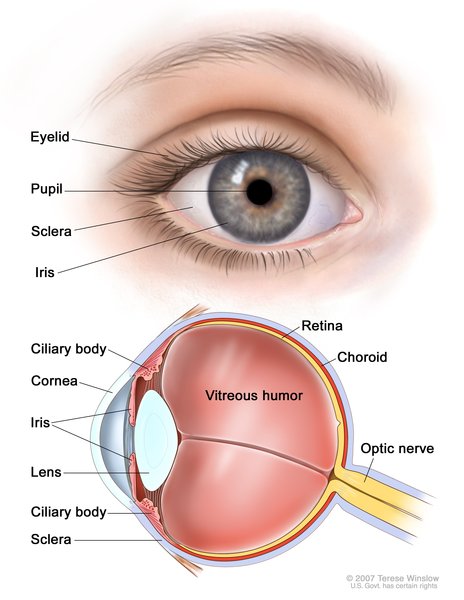
Children with Saul-Wilson syndrome often have hearing loss, clouding of the lenses of the eyes (cataracts), or a blue tint to the whites of the eyes (blue sclerae). They may also have retinitis pigmentosa, in which breakdown of the light-sensitive layer (retina) at the back of the eye can cause vision loss. Individuals with Saul-Wilson syndrome may have early delay of speech and motor development, but they usually have normal intelligence.
In Saul-Wilson syndrome, levels of white blood cells can vary from normal to low (intermittent neutropenia). Neutropenia makes it more difficult for the body to fight off foreign invaders such as bacteria and viruses, and may contribute to recurrent respiratory infections that occur in childhood in some individuals with Saul-Wilson syndrome.
Frequency
Saul-Wilson syndrome is a very rare disorder. At least 16 affected individuals have been reported in the scientific literature.
Causes
Saul-Wilson syndrome is caused by mutations in the COG4 gene. This gene provides instructions for making one piece of a group of proteins known as the conserved oligomeric Golgi (COG) complex. This complex functions in the Golgi apparatus, which is a cellular structure in which newly produced proteins are modified so they can carry out their functions. The COG complex plays an important role in the transport of proteins from the Golgi apparatus to another cellular structure called the endoplasmic reticulum. The endoplasmic reticulum processes proteins and helps move them to other structures in the cell. Transporting proteins from the Golgi apparatus to the endoplasmic reticulum (known as retrograde transport) is important for recycling proteins and relocating misplaced proteins.
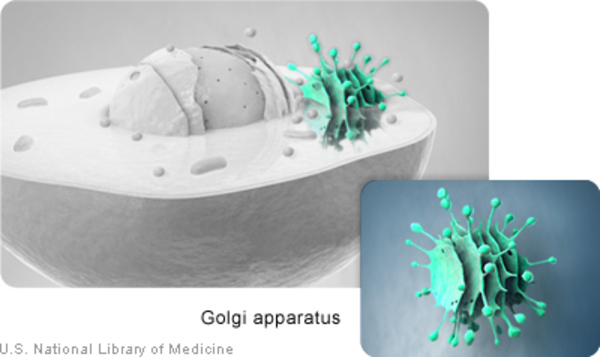

The COG4 gene mutations that cause Saul-Wilson syndrome result in production of an abnormal COG4 protein. Although the protein is altered, it is still able to be a part of the COG complex. When the abnormal COG4 protein is part of the COG complex, the transport of proteins between the Golgi apparatus and the endoplasmic reticulum is increased. It is unclear how this change in retrograde transport impairs bone growth and leads to the signs and symptoms of Saul-Wilson syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- Microcephalic osteodysplastic dysplasia
- Microcephalic osteodysplastic dysplasia Saul Wilson type
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Blackburn JB, D'Souza Z, Lupashin VV. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019 Sep;593(17):2466-2487. doi: 10.1002/1873-3468.13570. Epub 2019 Aug 16. Citation on PubMed or Free article on PubMed Central
- Ferreira C. Saul-Wilson Syndrome. 2020 Feb 20. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK554080/ Citation on PubMed
- Ferreira CR, Xia ZJ, Clement A, Parry DA, Davids M, Taylan F, Sharma P, Turgeon CT, Blanco-Sanchez B, Ng BG, Logan CV, Wolfe LA, Solomon BD, Cho MT, Douglas G, Carvalho DR, Bratke H, Haug MG, Phillips JB, Wegner J, Tiemeyer M, Aoki K; Undiagnosed Diseases Network; Scottish Genome Partnership; Nordgren A, Hammarsjo A, Duker AL, Rohena L, Hove HB, Ek J, Adams D, Tifft CJ, Onyekweli T, Weixel T, Macnamara E, Radtke K, Powis Z, Earl D, Gabriel M, Russi AHS, Brick L, Kozenko M, Tham E, Raymond KM, Phillips JA 3rd, Tiller GE, Wilson WG, Hamid R, Malicdan MCV, Nishimura G, Grigelioniene G, Jackson A, Westerfield M, Bober MB, Gahl WA, Freeze HH. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am J Hum Genet. 2018 Oct 4;103(4):553-567. doi: 10.1016/j.ajhg.2018.09.003. Citation on PubMed or Free article on PubMed Central
- Ferreira CR, Zein WM, Huryn LA, Merker A, Berger SI, Wilson WG, Tiller GE, Wolfe LA, Merideth M, Carvalho DR, Duker AL, Bratke H, Haug MG, Rohena L, Hove HB, Xia ZJ, Ng BG, Freeze HH, Gabriel M, Russi AHS, Brick L, Kozenko M, Earl DL, Tham E, Nishimura G, Phillips JA 3rd, Gahl WA, Hamid R, Jackson AP, Grigelioniene G, Bober MB. Defining the clinical phenotype of Saul-Wilson syndrome. Genet Med. 2020 May;22(5):857-866. doi: 10.1038/s41436-019-0737-1. Epub 2020 Jan 17. Citation on PubMed
- Saul RA, Wilson WG. COMMENTARY-The Saul-Wilson syndrome from its early days until now. Am J Med Genet A. 2019 Feb;179(2):159-160. doi: 10.1002/ajmg.a.8. Epub 2018 Dec 13. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.