

Description
RRM2B-related mitochondrial DNA depletion syndrome, encephalomyopathic form with renal tubulopathy (RRM2B-MDS) is a severe condition that begins in infancy and affects multiple body systems. It is associated with brain dysfunction combined with muscle weakness (encephalomyopathy). Many affected individuals also have a kidney dysfunction known as renal tubulopathy.

Infants with RRM2B-MDS have weak muscle tone (hypotonia) and a failure to grow or gain weight at the expected rate (failure to thrive). Many have a smaller-than-normal head size (microcephaly). Due to muscle weakness, affected infants typically have difficulty controlling head movement and may have delayed development of other motor skills, such as rolling over or sitting. Weakness of the muscles used for breathing leads to serious breathing difficulties and can result in life-threatening respiratory failure. Most affected infants have a buildup of a chemical called lactic acid in the body (lactic acidosis), which can also be life-threatening.
Some individuals with RRM2B-MDS have a digestion problem known as gastrointestinal dysmotility, in which the muscles and nerves of the digestive system do not move food through the digestive tract efficiently. This disorder may lead to swallowing difficulties, vomiting, and diarrhea and can contribute to a failure to thrive. Less commonly, individuals with RRM2B-MDS develop seizures or hearing loss that is caused by nerve damage in the inner ear (sensorineural hearing loss).

Because of the severity of the signs and symptoms, people with RRM2B-MDS usually live only into early childhood.
Frequency
RRM2B-MDS is a rare condition; the exact prevalence is unknown. At least 15 cases have been reported in the medical literature.
Causes
As the condition name suggests, mutations in the RRM2B gene cause RRM2B-MDS. The RRM2B gene provides instructions for making one piece, called the p53 inducible small subunit (p53R2), of a protein called ribonucleotide reductase (RNR). RNR helps produce DNA building blocks (nucleotides), which are joined to one another in a particular order to form DNA.
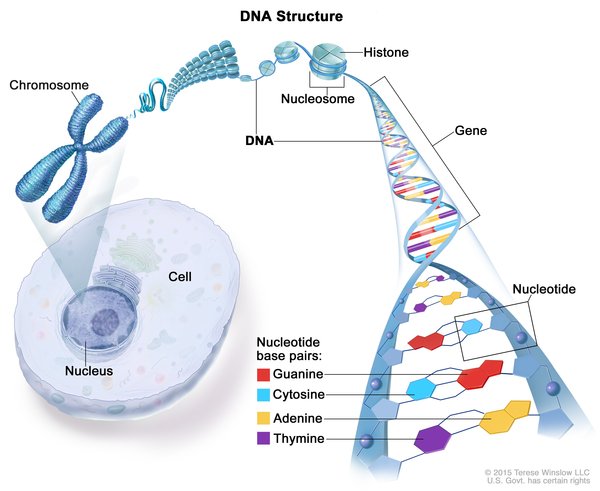
RNRs containing p53R2 make nucleotides that are used for the formation of DNA in specialized cell structures called mitochondria. Although most DNA is packaged in chromosomes within the cell's nucleus (nuclear DNA), mitochondria also have a small amount of their own DNA (mitochondrial DNA or mtDNA). Mitochondria are the energy-producing centers in cells, and the DNA in these structures contains genes essential for the process of energy production (called oxidative phosphorylation). The production of nucleotides by p53R2 helps maintain a normal amount of mtDNA in cells.

RRM2B gene mutations reduce the activity or amount of RNR, which likely impairs production of mtDNA nucleotides. A shortage of nucleotides available for the production of mtDNA molecules leads to a reduction in the amount of mtDNA (known as mtDNA depletion), which impairs mitochondrial function.
It is unclear why mtDNA depletion particularly affects the brain and kidneys in people with RRM2B-MDS, but the high energy demands of these tissues may make them especially susceptible to cell death when mtDNA is lost and less energy is produced in cells.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Mitochondrial DNA depletion syndrome 8A (encephalomyopathic type with renal tubulopathy)
- MTDPS8A
- RRM2B-MDS
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bornstein B, Area E, Flanigan KM, Ganesh J, Jayakar P, Swoboda KJ, Coku J, Naini A, Shanske S, Tanji K, Hirano M, DiMauro S. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord. 2008 Jun;18(6):453-9. doi: 10.1016/j.nmd.2008.04.006. Epub 2008 May 27. Citation on PubMed or Free article on PubMed Central
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007 Jun;39(6):776-80. doi: 10.1038/ng2040. Epub 2007 May 7. Citation on PubMed
- Lim AZ, McFarland R, Taylor RW, Gorman GS. RRM2B Mitochondrial DNA Maintenance Defects. 2014 Apr 17 [updated 2021 Jun 24]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK195854/ Citation on PubMed
- Pontarin G, Ferraro P, Bee L, Reichard P, Bianchi V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc Natl Acad Sci U S A. 2012 Aug 14;109(33):13302-7. doi: 10.1073/pnas.1211289109. Epub 2012 Jul 30. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.