

Description
Romano-Ward syndrome is a condition that causes a disruption of the heart's normal rhythm (arrhythmia). This disorder is a form of long QT syndrome, which is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The term "long QT" refers to a specific pattern of heart activity that is detected with an electrocardiogram (ECG or EKG), which is a test used to measure the electrical activity of the heart. In people with long QT syndrome, the part of the heartbeat known as the QT interval is abnormally long. Abnormalities in the time it takes to recharge the heart lead to abnormal heart rhythms.
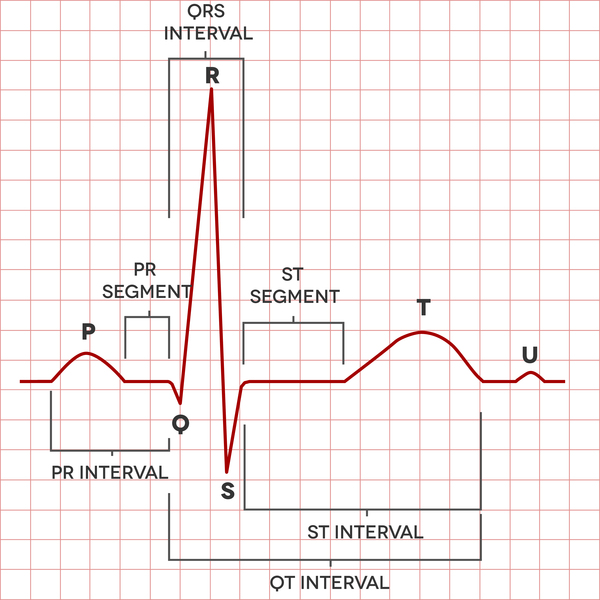
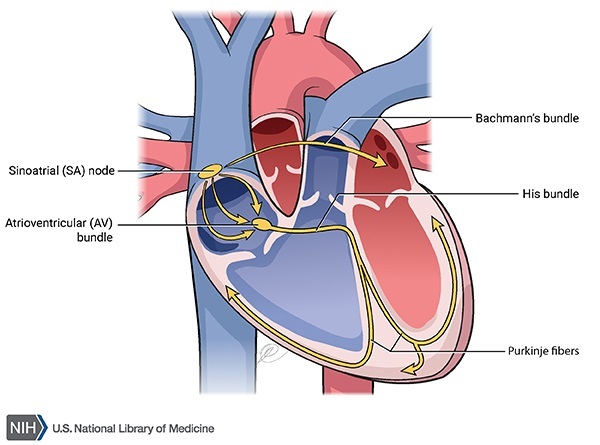
The arrhythmia associated with Romano-Ward syndrome can lead to fainting (syncope) or cardiac arrest and sudden death. However, some people with Romano-Ward syndrome never experience any health problems associated with the condition.
Fifteen types of long QT syndrome have been defined based on their genetic cause. Some types of long QT syndrome involve other cardiac abnormalities or problems with additional body systems. Romano-Ward syndrome encompasses those types that involve only a long QT interval without other abnormalities.
Frequency
Romano-Ward syndrome is the most common form of inherited long QT syndrome, which affects an estimated 1 in 2,000 people worldwide. Long QT syndrome may actually be more common than this estimate, however, because some people never experience any symptoms associated with arrhythmia and therefore may not be diagnosed.
Causes
Mutations in the KCNQ1, KCNH2, and SCN5A genes are the most common causes of Romano-Ward syndrome. These genes provide instructions for making proteins that form channels across the cell membrane. These channels transport positively charged atoms (ions), such as potassium and sodium, into and out of cells. In cardiac muscle cells, these ion channels play critical roles in maintaining the heart's normal rhythm. Mutations in any of these genes alter the structure or function of the channels, which changes the flow of ions in and out of cells. A disruption in ion transport alters the way the heart beats, leading to the abnormal heart rhythm characteristic of Romano-Ward syndrome.

Mutations in other genes involved in ion transport can also cause Romano-Ward syndrome; each of these additional genes is associated with a very small percentage of cases.
Inheritance
Romano-Ward syndrome follows an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the mutation from one affected parent. A small percentage of cases result from new mutations in one of the associated genes. These cases occur in people with no history of Romano-Ward syndrome in their family. Some people who have an altered gene never develop signs and symptoms of the condition, a situation known as reduced penetrance.


Other Names for This Condition
- RWS
- Ward-Romano syndrome
- WRS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol Rev. 2017 Jan;97(1):89-134. doi: 10.1152/physrev.00008.2016. Citation on PubMed
- Groffen AJ, Bikker H, Christiaans I. Long QT Syndrome Overview. 2003 Feb 20 [updated 2024 Mar 21]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1129/ Citation on PubMed
- Modell SM, Bradley DJ, Lehmann MH. Genetic testing for long QT syndrome and the category of cardiac ion channelopathies. PLoS Curr. 2012 May 3;4:e4f9995f69e6c7. doi: 10.1371/4f9995f69e6c7. Citation on PubMed or Free article on PubMed Central
- Nakano Y, Shimizu W. Genetics of long-QT syndrome. J Hum Genet. 2016 Jan;61(1):51-5. doi: 10.1038/jhg.2015.74. Epub 2015 Jun 25. Citation on PubMed
- Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012 Aug 1;5(4):868-77. doi: 10.1161/CIRCEP.111.962019. No abstract available. Citation on PubMed or Free article on PubMed Central
- Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 2014 Jan-Mar;10(1):29-33. doi: 10.14797/mdcj-10-1-29. Citation on PubMed or Free article on PubMed Central
- Towbin JA, Vatta M. Molecular biology and the prolonged QT syndromes. Am J Med. 2001 Apr 1;110(5):385-98. doi: 10.1016/s0002-9343(00)00715-4. Citation on PubMed
- Vincent GM. The Long QT and Brugada syndromes: causes of unexpected syncope and sudden cardiac death in children and young adults. Semin Pediatr Neurol. 2005 Mar;12(1):15-24. doi: 10.1016/j.spen.2004.11.008. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.