

Description
Rhizomelic chondrodysplasia punctata is a condition that impairs the normal development of many parts of the body. The major features of this disorder include skeletal abnormalities, distinctive facial features, intellectual disability, and respiratory problems.
Rhizomelic chondrodysplasia punctata is characterized by shortening of the bones in the upper arms and thighs (rhizomelia). Affected individuals also have a specific bone abnormality called chondrodysplasia punctata, which affects the growth of the long bones and can be seen on x-rays. People with rhizomelic chondrodysplasia punctata often develop joint deformities (contractures) that make the joints stiff and painful.
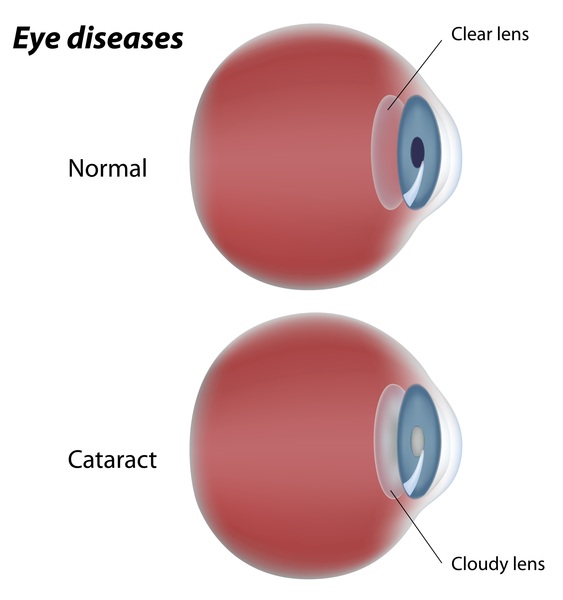
Distinctive facial features are also seen with rhizomelic chondrodysplasia punctata. These include a prominent forehead, widely set eyes (hypertelorism), a sunken appearance of the middle of the face (midface hypoplasia), a small nose with upturned nostrils, and full cheeks. Additionally, almost all affected individuals have clouding of the lenses of the eyes (cataracts). The cataracts are apparent at birth (congenital) or develop in early infancy.
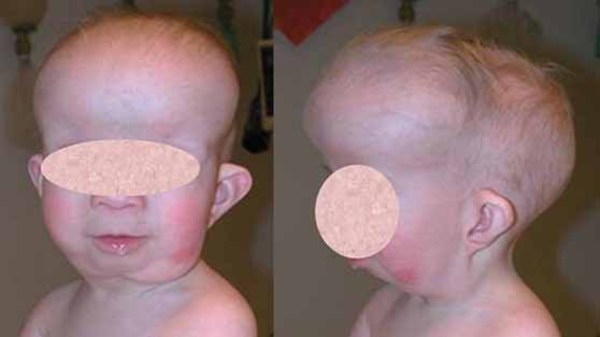

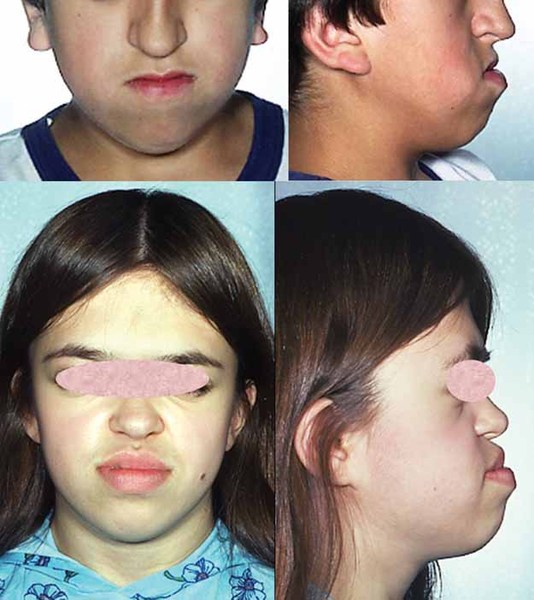
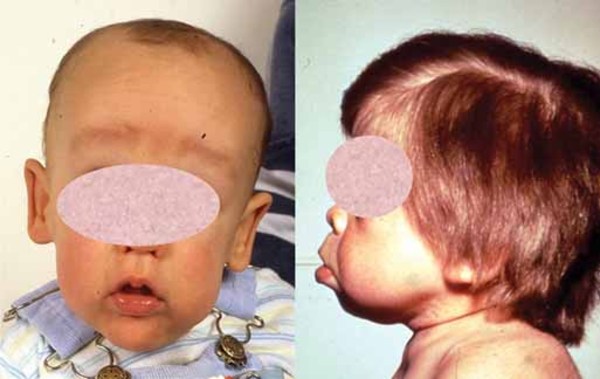
Rhizomelic chondrodysplasia punctata is associated with significantly delayed development and severe intellectual disability. Most children with this condition do not achieve developmental milestones such as sitting without support, feeding themselves, or speaking in phrases. Affected infants grow much more slowly than other children their age, and many also have seizures. Recurrent respiratory infections and life-threatening breathing problems are common. Because of their severe health problems, most people with rhizomelic chondrodysplasia punctata survive only into childhood. It is rare for affected children to live past age 10. However, a few individuals with milder features of the condition have lived into early adulthood.
Researchers have described three types of rhizomelic chondrodysplasia punctata: type 1 (RCDP1), type 2 (RCDP2), and type 3 (RCDP3). The types have similar features and are distinguished by their genetic cause.
Frequency
Rhizomelic chondrodysplasia punctata affects fewer than 1 in 100,000 people worldwide. RCDP1 is more common than RCDP2 or RCDP3.
Causes
Rhizomelic chondrodysplasia punctata results from mutations in one of three genes. Mutations in the PEX7 gene, which are most common, cause RCDP1. Changes in the GNPAT gene lead to RCDP2, while AGPS gene mutations result in RCDP3.
The genes associated with rhizomelic chondrodysplasia punctata are involved in the formation and function of structures called peroxisomes. Peroxisomes are sac-like compartments within cells that contain enzymes needed to break down many different substances, including fatty acids and certain toxic compounds. They are also important for the production of fats (lipids) used in digestion and in the nervous system.

Within peroxisomes, the proteins produced from the PEX7, GNPAT, and AGPS genes play roles in the formation (synthesis) of lipid molecules called plasmalogens. Plasmalogens are found in cell membranes throughout the body, although little is known about their function. Mutations in the PEX7, GNPAT, or AGPS genes prevent peroxisomes from making plasmalogens. Researchers are working to determine how problems with plasmalogen synthesis lead to the specific signs and symptoms of rhizomelic chondrodysplasia punctata.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Chondrodysplasia punctata, rhizomelic
- RCDP
- RCP
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Braverman NE, Carroll R, Muss C, Fallatah W, Jain M. PEX7-Related Rhizomelic Chondrodysplasia Punctata. 2001 Nov 16 [updated 2025 Aug 7]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1270/ Citation on PubMed
- Irving MD, Chitty LS, Mansour S, Hall CM. Chondrodysplasia punctata: a clinical diagnostic and radiological review. Clin Dysmorphol. 2008 Oct;17(4):229-41. doi: 10.1097/MCD.0b013e3282fdcc70. Citation on PubMed
- Phadke SR, Gupta N, Girisha KM, Kabra M, Maeda M, Vidal E, Moser A, Steinberg S, Puri RD, Verma IC, Braverman N. Rhizomelic chondrodysplasia punctata type 1: report of mutations in 3 children from India. J Appl Genet. 2010;51(1):107-10. doi: 10.1007/BF03195717. Citation on PubMed
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006 Dec;1763(12):1733-48. doi: 10.1016/j.bbamcr.2006.09.010. Epub 2006 Sep 14. Citation on PubMed
- White AL, Modaff P, Holland-Morris F, Pauli RM. Natural history of rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2003 May 1;118A(4):332-42. doi: 10.1002/ajmg.a.20009. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.