

Description
Pyle disease is a disorder of the bones. Its hallmark feature is an abnormality of the long bones in the arms and legs in which the ends (metaphyses) of the bones are abnormally broad; the shape of the bones resembles a boat oar or paddle. The broad metaphyses are due to enlargement of the spongy inner layer of bone (trabecular bone). Although trabecular bone is expanded, the dense outermost layer of bone (cortical bone) is thinner than normal. As a result, the bones are fragile and fracture easily. The bone abnormalities in the legs commonly cause knock knees (genu valgum) in affected individuals.
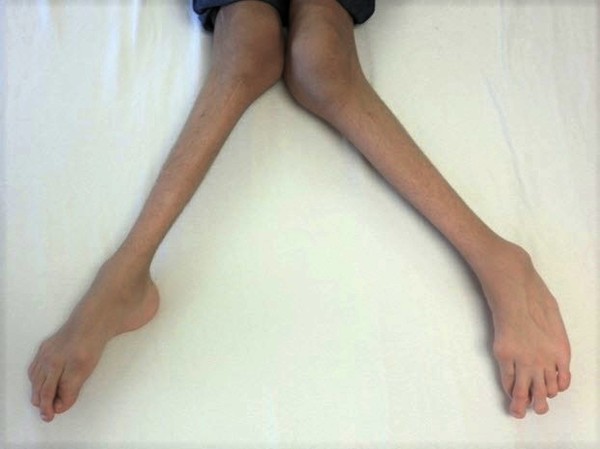
Other bone abnormalities can also occur in Pyle disease. Affected individuals may have widened collar bones (clavicles), ribs, or bones in the fingers and hands. Dental problems are common in Pyle disease, including delayed appearance (eruption) of permanent teeth and misalignment of the top and bottom teeth (malocclusion).
Frequency
Pyle disease is thought to be a rare disorder, although its prevalence is unknown. More than 25 cases have been described in the medical literature.
Causes
Pyle disease is caused by mutations in the SFRP4 gene. This gene provides instructions for making a protein that blocks (inhibits) a process called Wnt signaling, which is involved in the development of several tissues and organs throughout the body. In particular, regulation of Wnt signaling by the SFRP4 protein is critical for normal bone development and remodeling. Bone remodeling is a normal process in which old bone is broken down and new bone is created to replace it. Mutations in the SFRP4 gene are thought to prevent the production of functional SFRP4 protein. The resulting dysregulation of Wnt signaling leads to the bone abnormalities characteristic of Pyle disease.
Inheritance
Pyle disease is inherited in an autosomal recessive pattern, which means both copies of the SFRP4 gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene. While they do not develop the condition, they may have mild abnormalities of the long bones.

Other Names for This Condition
- Metaphyseal dysplasia, Pyle type
- Pyle metaphyseal dysplasia
- Pyle's disease
- Pyle's metaphyseal dysplasia syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Galada C, Shah H, Shukla A, Girisha KM. A novel sequence variant in SFRP4 causing Pyle disease. J Hum Genet. 2017 Apr;62(5):575-576. doi: 10.1038/jhg.2016.166. Epub 2017 Jan 19. Citation on PubMed
- Haraguchi R, Kitazawa R, Mori K, Tachibana R, Kiyonari H, Imai Y, Abe T, Kitazawa S. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci Rep. 2016 Apr 27;6:25198. doi: 10.1038/srep25198. Citation on PubMed or Free article on PubMed Central
- Kiper POS, Saito H, Gori F, Unger S, Hesse E, Yamana K, Kiviranta R, Solban N, Liu J, Brommage R, Boduroglu K, Bonafe L, Campos-Xavier B, Dikoglu E, Eastell R, Gossiel F, Harshman K, Nishimura G, Girisha KM, Stevenson BJ, Takita H, Rivolta C, Superti-Furga A, Baron R. Cortical-Bone Fragility--Insights from sFRP4 Deficiency in Pyle's Disease. N Engl J Med. 2016 Jun 30;374(26):2553-2562. doi: 10.1056/NEJMoa1509342. Citation on PubMed or Free article on PubMed Central
- Wonkam A, Makubalo N, Roberts T, Chetty M. Pyle metaphyseal dysplasia in an African child: Case report and review of the literature. S Afr Med J. 2016 May 25;106(6 Suppl 1):S110-3. doi: 10.7196/SAMJ.2016.v106i6.11011. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.