

Description
Prolidase deficiency is a disorder that causes a wide variety of symptoms. The disorder typically becomes apparent during infancy. Affected individuals may have enlargement of the spleen (splenomegaly); in some cases, both the spleen and liver are enlarged (hepatosplenomegaly). Diarrhea, vomiting, and dehydration may also occur. People with prolidase deficiency are susceptible to severe infections of the skin or ears, or potentially life-threatening respiratory tract infections. Some individuals with prolidase deficiency have chronic lung disease.
Characteristic facial features in people with prolidase deficiency include prominent eyes that are widely spaced (hypertelorism), a high forehead, a flat bridge of the nose, and a very small lower jaw and chin (micrognathia). Affected children may experience delayed development, and about 75 percent of people with prolidase deficiency have intellectual disability that may range from mild to severe.
People with prolidase deficiency often develop skin lesions, especially on their hands, feet, lower legs, and face. The severity of the skin involvement, which usually begins during childhood, may range from a mild rash to severe skin ulcers. Skin ulcers, especially on the legs, may not heal completely, resulting in complications including infection and amputation.
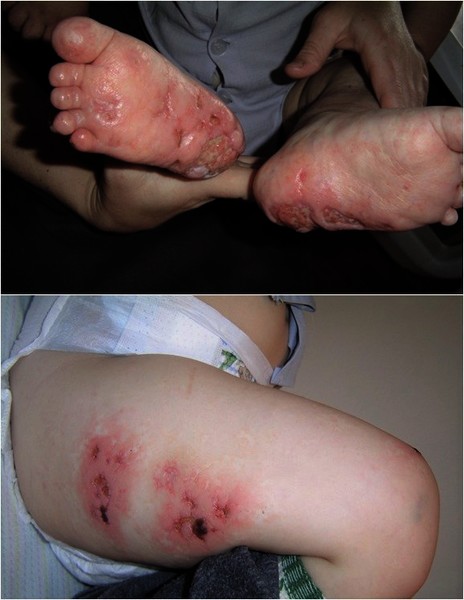
The severity of symptoms in prolidase deficiency varies greatly among affected individuals. Some people with this disorder do not have any symptoms. In these individuals the condition can be detected by laboratory tests such as newborn screening tests or tests offered to relatives of affected individuals.
Frequency
Prolidase deficiency is a rare disorder. Approximately 70 individuals with this disorder have been documented in the medical literature, and researchers have estimated that the condition occurs in approximately 1 in 1 million to 1 in 2 million newborns. It is more common in certain areas in northern Israel, both among members of a religious minority called the Druze and in nearby Arab Moslem populations.
Causes
Prolidase deficiency is caused by mutations in the PEPD gene. This gene provides instructions for making the enzyme prolidase, also called peptidase D. Prolidase helps divide certain dipeptides, which are molecules composed of two protein building blocks (amino acids). Specifically, prolidase divides dipeptides containing the amino acids proline or hydroxyproline. By freeing these amino acids, prolidase helps make them available for use in producing proteins that the body needs.

Prolidase is also involved in the final step of the breakdown of some proteins obtained through the diet and proteins that are no longer needed in the body. Prolidase is particularly important in the breakdown of collagens, a family of proteins that are rich in proline and hydroxyproline. Collagens are an important part of the extracellular matrix, which is the lattice of proteins and other molecules outside the cell. The extracellular matrix strengthens and supports connective tissues, such as skin, bone, cartilage, tendons, and ligaments. Collagen breakdown occurs during the maintenance (remodeling) of the extracellular matrix.
PEPD gene mutations that cause prolidase deficiency result in the loss of prolidase enzyme activity. It is not well understood how the absence of prolidase activity causes the various signs and symptoms of prolidase deficiency. Researchers have suggested that accumulation of dipeptides that have not been broken down may lead to cell death. When cells die, their contents are released into the surrounding tissue, which could cause inflammation and lead to the skin problems seen in prolidase deficiency. Impaired collagen breakdown during remodeling of the extracellular matrix may also contribute to the skin problems. The intellectual disability that occurs in prolidase deficiency might result from problems in processing neuropeptides, which are brain signaling proteins that are rich in proline. It is unclear how absence of prolidase activity results in the other features of prolidase deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Hyperimidodipeptiduria
- Imidodipeptidase deficiency
- PD
- Peptidase deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Falik-Zaccai TC, Khayat M, Luder A, Frenkel P, Magen D, Brik R, Gershoni-Baruch R, Mandel H. A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am J Med Genet B Neuropsychiatr Genet. 2010 Jan 5;153B(1):46-56. doi: 10.1002/ajmg.b.30945. Citation on PubMed
- Forlino A, Lupi A, Vaghi P, Icaro Cornaglia A, Calligaro A, Campari E, Cetta G. Mutation analysis of five new patients affected by prolidase deficiency: the lack of enzyme activity causes necrosis-like cell death in cultured fibroblasts. Hum Genet. 2002 Oct;111(4-5):314-22. doi: 10.1007/s00439-002-0792-5. Epub 2002 Aug 14. Citation on PubMed
- Luder AS, Mandel H, Khayat M, Gurevich I, Frankel P, Rivlin J, Falik-Zaccai TC. Chronic lung disease and cystic fibrosis phenotype in prolidase deficiency: a newly recognized association. J Pediatr. 2007 Jun;150(6):656-8, 658.e1. doi: 10.1016/j.jpeds.2007.03.025. Citation on PubMed
- Lupi A, De Riso A, Torre SD, Rossi A, Campari E, Vilarinho L, Cetta G, Forlino A. Characterization of a new PEPD allele causing prolidase deficiency in two unrelated patients: natural-occurrent mutations as a tool to investigate structure-function relationship. J Hum Genet. 2004;49(9):500-506. doi: 10.1007/s10038-004-0180-1. Epub 2004 Aug 11. Citation on PubMed
- Lupi A, Rossi A, Campari E, Pecora F, Lund AM, Elcioglu NH, Gultepe M, Di Rocco M, Cetta G, Forlino A. Molecular characterisation of six patients with prolidase deficiency: identification of the first small duplication in the prolidase gene and of a mutation generating symptomatic and asymptomatic outcomes within the same family. J Med Genet. 2006 Dec;43(12):e58. doi: 10.1136/jmg.2006.043315. Citation on PubMed or Free article on PubMed Central
- Lupi A, Tenni R, Rossi A, Cetta G, Forlino A. Human prolidase and prolidase deficiency: an overview on the characterization of the enzyme involved in proline recycling and on the effects of its mutations. Amino Acids. 2008 Nov;35(4):739-52. doi: 10.1007/s00726-008-0055-4. Epub 2008 Mar 14. Citation on PubMed
- Mitsubuchi H, Nakamura K, Matsumoto S, Endo F. Inborn errors of proline metabolism. J Nutr. 2008 Oct;138(10):2016S-2020S. doi: 10.1093/jn/138.10.2016S. Citation on PubMed
- Wang H, Kurien BT, Lundgren D, Patel NC, Kaufman KM, Miller DL, Porter AC, D'Souza A, Nye L, Tumbush J, Hupertz V, Kerr DS, Kurono S, Matsumoto H, Scofield RH. A nonsense mutation of PEPD in four Amish children with prolidase deficiency. Am J Med Genet A. 2006 Mar 15;140(6):580-5. doi: 10.1002/ajmg.a.31134. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.