

Description
Pompe disease is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body's cells. The accumulation of glycogen in certain organs and tissues, especially muscles, impairs their ability to function normally.
Researchers have described three types of Pompe disease, which differ in severity and the age at which they appear. These types are known as classic infantile-onset, non-classic infantile-onset, and late-onset.
The classic form of infantile-onset Pompe disease begins within a few months of birth. Infants with this disorder typically experience muscle weakness (myopathy), poor muscle tone (hypotonia), an enlarged liver (hepatomegaly), and heart defects. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems. If untreated, this form of Pompe disease leads to death from heart failure in the first year of life.
The non-classic form of infantile-onset Pompe disease usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
The late-onset type of Pompe disease may not become apparent until later in childhood, adolescence, or adulthood. Late-onset Pompe disease is usually milder than the infantile-onset forms of this disorder and is less likely to involve the heart. Most individuals with late-onset Pompe disease experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure.
Frequency
Pompe disease affects about 1 in 40,000 people in the United States. The incidence of this disorder varies among different ethnic groups.
Causes
Mutations in the GAA gene cause Pompe disease. The GAA gene provides instructions for producing an enzyme called acid alpha-glucosidase (also known as acid maltase). This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. The enzyme normally breaks down glycogen into a simpler sugar called glucose, which is the main energy source for most cells.
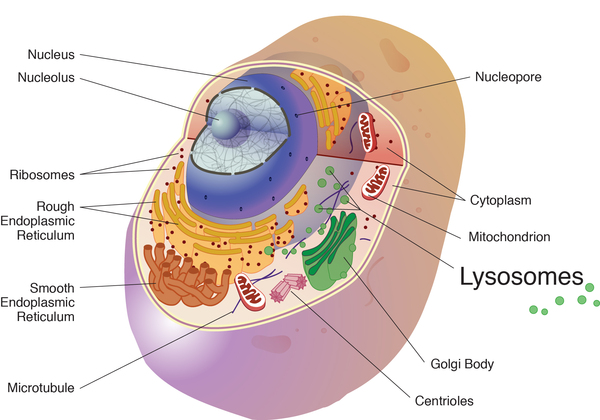
Mutations in the GAA gene prevent acid alpha-glucosidase from breaking down glycogen effectively, which allows this sugar to build up to toxic levels in lysosomes. This buildup damages organs and tissues throughout the body, particularly the muscles, leading to the progressive signs and symptoms of Pompe disease.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Acid maltase deficiency
- Acid maltase deficiency disease
- Alpha-1,4-glucosidase deficiency
- AMD
- Deficiency of alpha-glucosidase
- GAA deficiency
- Glycogen storage disease type II
- Glycogenosis type II
- GSD II
- GSD2
- Pompe's disease
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bembi B, Cerini E, Danesino C, Donati MA, Gasperini S, Morandi L, Musumeci O, Parenti G, Ravaglia S, Seidita F, Toscano A, Vianello A. Diagnosis of glycogenosis type II. Neurology. 2008 Dec 2;71(23 Suppl 2):S4-11. doi: 10.1212/WNL.0b013e31818da91e. Citation on PubMed
- Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, Huang AC, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009 Dec;124(6):e1116-25. doi: 10.1542/peds.2008-3667. Citation on PubMed
- Fukuda T, Roberts A, Plotz PH, Raben N. Acid alpha-glucosidase deficiency (Pompe disease). Curr Neurol Neurosci Rep. 2007 Jan;7(1):71-7. doi: 10.1007/s11910-007-0024-4. Citation on PubMed
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D; Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006 May;148(5):671-676. doi: 10.1016/j.jpeds.2005.11.033. Citation on PubMed
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O'Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006 May;8(5):267-88. doi: 10.1097/01.gim.0000218152.87434.f3. No abstract available. Citation on PubMed or Free article on PubMed Central
- van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003 Aug;112(2):332-40. doi: 10.1542/peds.112.2.332. Citation on PubMed
- van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008 Oct 11;372(9646):1342-53. doi: 10.1016/S0140-6736(08)61555-X. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.