

Description
PMM2-congenital disorder of glycosylation (PMM2-CDG, also known as congenital disorder of glycosylation type Ia) is an inherited condition that affects many parts of the body. The type and severity of problems associated with PMM2-CDG vary widely among affected individuals, sometimes even among members of the same family.
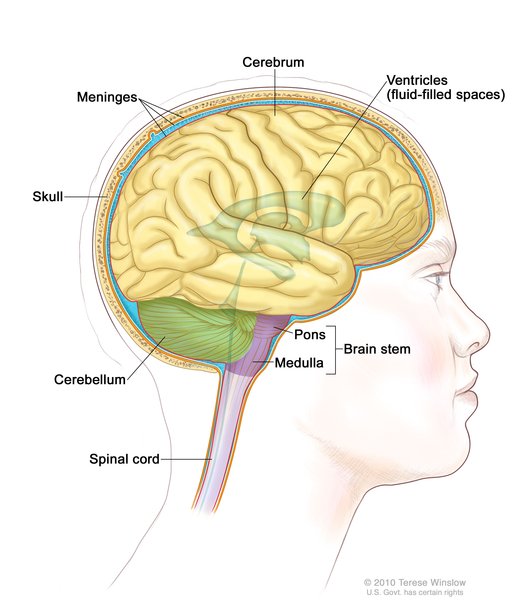
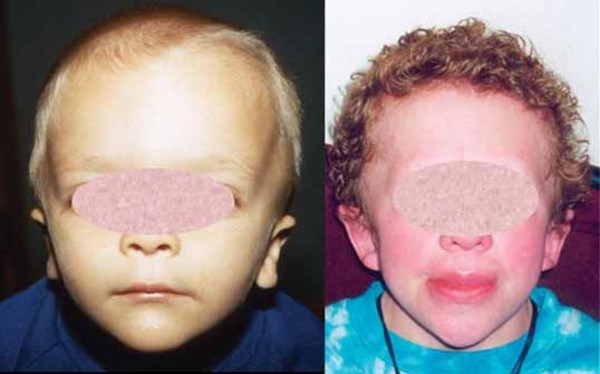
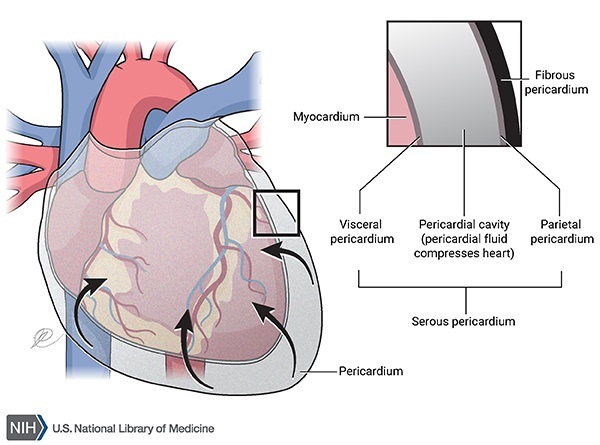
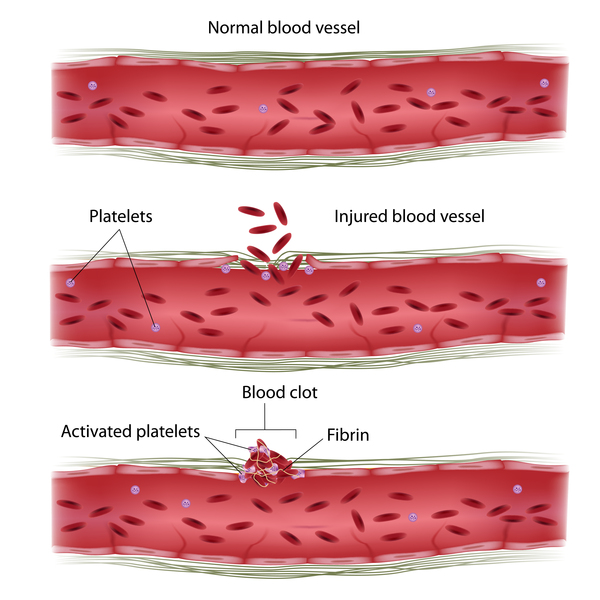
Individuals with PMM2-CDG typically develop signs and symptoms of the condition during infancy. Affected infants may have weak muscle tone (hypotonia), retracted (inverted) nipples, an abnormal distribution of fat, eyes that do not look in the same direction (strabismus), developmental delay, and a failure to gain weight and grow at the expected rate (failure to thrive). Infants with PMM2-CDG also frequently have an underdeveloped cerebellum, which is the part of the brain that coordinates movement. Distinctive facial features are sometimes present in affected individuals, including a high forehead, a triangular face, large ears, and a thin upper lip. Children with PMM2-CDG may also have elevated liver function test results, seizures, fluid around the heart (pericardial effusion), and blood clotting disorders. About 20 percent of affected infants do not survive the first year of life due to multiple organ failure.

The most severe cases of PMM2-CDG are characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. Most babies with hydrops fetalis are stillborn or die soon after birth.
People with PMM2-CDG who survive infancy may have moderate intellectual disability, and some are unable to walk independently. Affected individuals may also experience stroke-like episodes that involve an extreme lack of energy (lethargy) and temporary paralysis. Recovery from these episodes usually occurs over a period of a few weeks to several months.
During adolescence or adulthood, individuals with PMM2-CDG have reduced sensation and weakness in their arms and legs (peripheral neuropathy), an abnormal curvature of the spine (kyphoscoliosis), impaired muscle coordination (ataxia), and joint deformities (contractures). Some affected individuals have an eye disorder called retinitis pigmentosa that causes vision loss. Females with PMM2-CDG have hypergonadotropic hypogonadism, which affects the production of hormones that direct sexual development. As a result, females with PMM2-CDG do not go through puberty. Affected males experience normal puberty but often have small testes.
Frequency
More than 800 individuals with PMM2-CDG have been identified worldwide.
Causes
PMM2-CDG is caused by mutations in the PMM2 gene. This gene provides instructions for making an enzyme called phosphomannomutase 2 (PMM2). The PMM2 enzyme is involved in a process called glycosylation, which attaches groups of sugar molecules (oligosaccharides) to proteins. Glycosylation modifies proteins so they can perform a wider variety of functions. Mutations in the PMM2 gene lead to the production of an abnormal PMM2 enzyme with reduced activity. Without a properly functioning PMM2 enzyme, glycosylation cannot proceed normally. As a result, incorrect oligosaccharides are produced and attached to proteins. The wide variety of signs and symptoms in PMM2-CDG are likely due to the production of abnormally glycosylated proteins in many organs and tissues.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Carbohydrate-deficient glycoprotein syndrome type Ia
- CDG Ia
- CDG1a
- CDGS1a
- Congenital disorder of glycosylation type Ia
- Jaeken syndrome
- Phosphomannomutase 2 deficiency
- PMM deficiency
- PMM2-CDG
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- de Lonlay P, Seta N, Barrot S, Chabrol B, Drouin V, Gabriel BM, Journel H, Kretz M, Laurent J, Le Merrer M, Leroy A, Pedespan D, Sarda P, Villeneuve N, Schmitz J, van Schaftingen E, Matthijs G, Jaeken J, Korner C, Munnich A, Saudubray JM, Cormier-Daire V. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001 Jan;38(1):14-9. doi: 10.1136/jmg.38.1.14. Citation on PubMed or Free article on PubMed Central
- Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta. 2009 Sep;1792(9):827-34. doi: 10.1016/j.bbadis.2009.01.003. Epub 2009 Jan 14. Citation on PubMed
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009 Dec;30(12):1628-41. doi: 10.1002/humu.21126. Citation on PubMed
- Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009 Sep;1792(9):825-6. doi: 10.1016/j.bbadis.2009.08.005. No abstract available. Citation on PubMed or Free article on PubMed Central
- Krasnewich D, O'Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia). Am J Med Genet C Semin Med Genet. 2007 Aug 15;145C(3):302-6. doi: 10.1002/ajmg.c.30143. Citation on PubMed
- Lam C, Krasnewich DM. PMM2-CDG. 2005 Aug 15 [updated 2021 May 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1110/ Citation on PubMed
- Monin ML, Mignot C, De Lonlay P, Heron B, Masurel A, Mathieu-Dramard M, Lenaerts C, Thauvin C, Gerard M, Roze E, Jacquette A, Charles P, de Barace C, Drouin-Garraud V, Khau Van Kien P, Cormier-Daire V, Mayer M, Ogier H, Brice A, Seta N, Heron D. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis. 2014 Dec 11;9:207. doi: 10.1186/s13023-014-0207-4. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.