

Description
Persistent Müllerian duct syndrome is a disorder of sexual development that affects males. Males with this disorder have normal male reproductive organs, though they also have a uterus and fallopian tubes, which are female reproductive organs. The uterus and fallopian tubes are derived from a structure called the Müllerian duct during development of the fetus. The Müllerian duct usually breaks down during early development in males, but it is retained in those with persistent Müllerian duct syndrome. Affected individuals have the normal chromosomes of a male (46,XY) and normal external male genitalia.
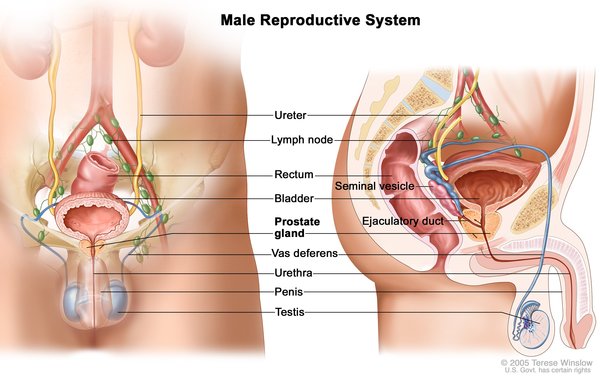
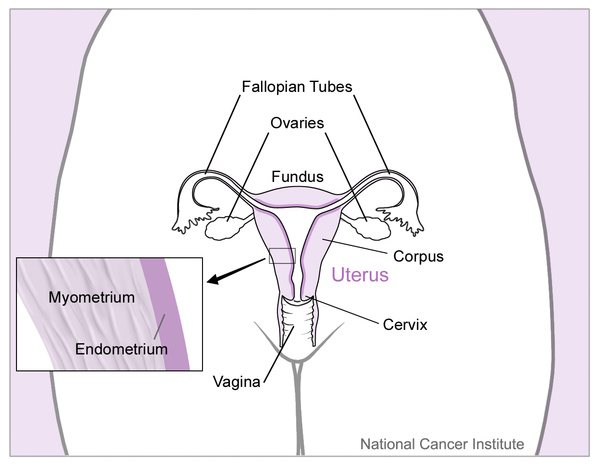
The first noted signs and symptoms in males with persistent Müllerian duct syndrome are usually undescended testes (cryptorchidism) or soft out-pouchings in the lower abdomen (inguinal hernias). The uterus and fallopian tubes are typically discovered when surgery is performed to treat these conditions.
The testes and female reproductive organs can be located in unusual positions in persistent Müllerian duct syndrome. Occasionally, both testes are undescended (bilateral cryptorchidism) and the uterus is in the pelvis. More often, one testis has descended into the scrotum normally, and one has not. Sometimes, the descended testis pulls the fallopian tube and uterus into the track through which it has descended. This creates a condition called hernia uteri inguinalis, a form of inguinal hernia. In other cases, the undescended testis from the other side of the body is also pulled into the same track, forming an inguinal hernia. This condition, called transverse testicular ectopia, is common in people with persistent Müllerian duct syndrome.
Other effects of persistent Müllerian duct syndrome may include the inability to father children (infertility) or blood in the semen (hematospermia). Also, the undescended testes may break down (degenerate) or develop cancer if left untreated.
Frequency
Persistent Müllerian duct syndrome is a rare disorder; however, the prevalence of the condition is unknown.
Causes
Most people with persistent Müllerian duct syndrome have mutations in the AMH gene or the AMHR2 gene. The AMH gene provides instructions for making a protein called anti-Müllerian hormone (AMH). The AMHR2 gene provides instructions for making a protein called AMH receptor type 2.
The AMH protein and AMH receptor type 2 protein are involved in male sex differentiation. All fetuses develop the Müllerian duct, the precursor to female reproductive organs. During development of a male fetus, these two proteins work together to induce breakdown (regression) of the Müllerian duct. Mutations in the AMH and AMHR2 genes lead to nonfunctional proteins that cannot signal for regression of the Müllerian duct. As a result of these mutations, the Müllerian duct persists and goes on to form a uterus and fallopian tubes.
Approximately 45 percent of cases of persistent Müllerian duct syndrome are caused by mutations in the AMH gene and are called persistent Müllerian duct syndrome type 1. Approximately 40 percent of cases are caused by mutations in the AMHR2 gene and are called persistent Müllerian duct syndrome type 2. In the remaining 15 percent of cases, no mutations in the AMH and AMHR2 genes have been identified, and the genes involved in causing the condition are unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. However, persistent Müllerian duct syndrome affects only males. Females with two mutated copies of the gene do not show signs and symptoms of the condition.

Other Names for This Condition
- Persistent oviduct syndrome
- PMDS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Belville C, Van Vlijmen H, Ehrenfels C, Pepinsky B, Rezaie AR, Picard JY, Josso N, di Clemente N, Cate RL. Mutations of the anti-mullerian hormone gene in patients with persistent mullerian duct syndrome: biosynthesis, secretion, and processing of the abnormal proteins and analysis using a three-dimensional model. Mol Endocrinol. 2004 Mar;18(3):708-21. doi: 10.1210/me.2003-0358. Epub 2003 Dec 12. Citation on PubMed
- Faure E, Gouedard L, Imbeaud S, Cate R, Picard JY, Josso N, di Clemente N. Mutant isoforms of the anti-Mullerian hormone type II receptor are not expressed at the cell membrane. J Biol Chem. 1996 Nov 29;271(48):30571-5. doi: 10.1074/jbc.271.48.30571. Citation on PubMed
- Imbeaud S, Belville C, Messika-Zeitoun L, Rey R, di Clemente N, Josso N, Picard JY. A 27 base-pair deletion of the anti-mullerian type II receptor gene is the most common cause of the persistent mullerian duct syndrome. Hum Mol Genet. 1996 Sep;5(9):1269-77. doi: 10.1093/hmg/5.9.1269. Citation on PubMed
- Josso N, Belville C, di Clemente N, Picard JY. AMH and AMH receptor defects in persistent Mullerian duct syndrome. Hum Reprod Update. 2005 Jul-Aug;11(4):351-6. doi: 10.1093/humupd/dmi014. Epub 2005 May 5. Citation on PubMed
- Josso N, Picard JY, Imbeaud S, di Clemente N, Rey R. Clinical aspects and molecular genetics of the persistent mullerian duct syndrome. Clin Endocrinol (Oxf). 1997 Aug;47(2):137-44. doi: 10.1046/j.1365-2265.1997.2411044.x. No abstract available. Citation on PubMed
- Rey R. Anti-Mullerian hormone in disorders of sex determination and differentiation. Arq Bras Endocrinol Metabol. 2005 Feb;49(1):26-36. doi: 10.1590/s0004-27302005000100005. Epub 2006 Mar 16. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.