

Description
Pantothenate kinase-associated neurodegeneration (formerly called Hallervorden-Spatz syndrome) is a disorder of the nervous system. This condition is characterized by progressive difficulty with movement, typically beginning in childhood. Movement abnormalities include involuntary muscle spasms, rigidity, and trouble with walking that worsens over time. Many people with this condition also develop problems with speech (dysarthria), and some develop vision loss. Additionally, affected individuals may experience a loss of intellectual function (dementia) and psychiatric symptoms such as behavioral problems, personality changes, and depression.
Pantothenate kinase-associated neurodegeneration is characterized by an abnormal buildup of iron in certain areas of the brain. A particular change called the eye-of-the-tiger sign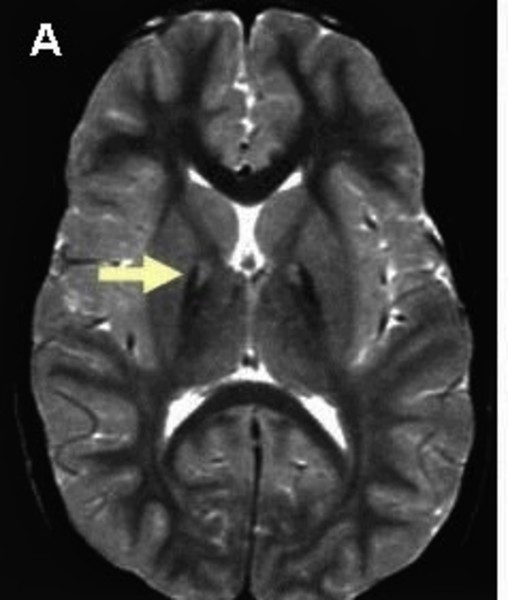 , which indicates an accumulation of iron, is typically seen on magnetic resonance imaging (MRI) scans of the brain in people with this disorder.
, which indicates an accumulation of iron, is typically seen on magnetic resonance imaging (MRI) scans of the brain in people with this disorder.
Researchers have described classic and atypical forms of pantothenate kinase-associated neurodegeneration. The classic form usually appears in early childhood, causing severe problems with movement that worsen rapidly. Features of the atypical form appear later in childhood or adolescence and progress more slowly. Signs and symptoms vary, but the atypical form is more likely than the classic form to involve speech defects and psychiatric problems.
A condition called HARP (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration) syndrome, which was historically described as a separate syndrome, is now considered part of pantothenate kinase-associated neurodegeneration.
Frequency
The precise incidence of this condition is unknown. It is estimated to affect 1 to 3 per million people worldwide.
Causes
Mutations in the PANK2 gene cause pantothenate kinase-associated neurodegeneration.
The PANK2 gene provides instructions for making an enzyme called pantothenate kinase 2. This enzyme is active in mitochondria, the energy-producing centers within cells, where it plays a critical role in the formation of a molecule called coenzyme A. Found in all living cells, coenzyme A is essential for the body's production of energy from carbohydrates, fats, and some protein building blocks (amino acids).
Mutations in the PANK2 gene likely result in the production of an abnormal version of pantothenate kinase 2 or prevent cells from making any of this enzyme. A lack of functional pantothenate kinase 2 disrupts the production of coenzyme A and allows potentially harmful compounds to build up in the brain. This buildup leads to swelling and tissue damage, and allows iron to accumulate abnormally in certain parts of the brain. Researchers have not determined how these changes result in the specific features of pantothenate kinase-associated neurodegeneration. Because pantothenate kinase 2 functions in mitochondria, the signs and symptoms of this condition may be related to impaired energy production.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- NBIA1
- Neurodegeneration with brain iron accumulation type 1
- PKAN
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Gregory A, Hayflick SJ. Neurodegeneration with brain iron accumulation. Folia Neuropathol. 2005;43(4):286-96. Citation on PubMed or Free article on PubMed Central
- Gregory A, Hayflick SJ. Pantothenate Kinase-Associated Neurodegeneration. 2002 Aug 13 [updated 2017 Aug 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1490/ Citation on PubMed
- Hartig MB, Hortnagel K, Garavaglia B, Zorzi G, Kmiec T, Klopstock T, Rostasy K, Svetel M, Kostic VS, Schuelke M, Botz E, Weindl A, Novakovic I, Nardocci N, Prokisch H, Meitinger T. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol. 2006 Feb;59(2):248-56. doi: 10.1002/ana.20771. Citation on PubMed
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, Gitschier J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003 Jan 2;348(1):33-40. doi: 10.1056/NEJMoa020817. Citation on PubMed
- Hayflick SJ. Pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). J Neurol Sci. 2003 Mar 15;207(1-2):106-7. doi: 10.1016/s0022-510x(02)00433-1. No abstract available. Citation on PubMed
- Hayflick SJ. Unraveling the Hallervorden-Spatz syndrome: pantothenate kinase-associated neurodegeneration is the name. Curr Opin Pediatr. 2003 Dec;15(6):572-7. doi: 10.1097/00008480-200312000-00005. Citation on PubMed
- Houlden H, Lincoln S, Farrer M, Cleland PG, Hardy J, Orrell RW. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology. 2003 Nov 25;61(10):1423-6. doi: 10.1212/01.wnl.0000094120.09977.92. Citation on PubMed
- Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, Scarano V, Bonuccelli U, Bentivoglio AR, D'Amico A, Marelli C, Di Giorgio A, Coubes P, Barone P, Dallapiccola B. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology. 2005 May 24;64(10):1810-2. doi: 10.1212/01.WNL.0000161843.52641.EC. Citation on PubMed
- Shevell M. Hallervorden and history. N Engl J Med. 2003 Jan 2;348(1):3-4. doi: 10.1056/NEJMp020158. No abstract available. Citation on PubMed
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001 Aug;28(4):345-9. doi: 10.1038/ng572. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.