

Description
The Maat-Kievit-Brunner type of Ohdo syndrome is a rare condition characterized by intellectual disability and distinctive facial features. It has only been reported in males.
The intellectual disability associated with this condition varies from mild to severe, and the development of motor skills (such as sitting, standing, and walking) is delayed. Some affected individuals also have behavioral problems.
Distinctive facial features often seen in this condition include a narrowing of the eye opening (blepharophimosis), droopy eyelids (ptosis), prominent cheeks, a broad nasal bridge, a nose with a rounded tip, a large space between the nose and upper lip (a long philtrum), and a narrow mouth. Some affected individuals also have widely set eyes (hypertelorism), an unusually small chin (micrognathia), and small and low-set ears. As people with the condition get older, these facial characteristics become more pronounced and the face becomes more triangular.
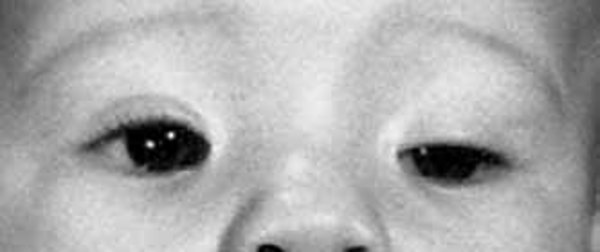
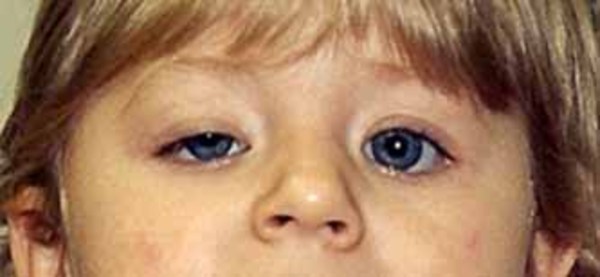
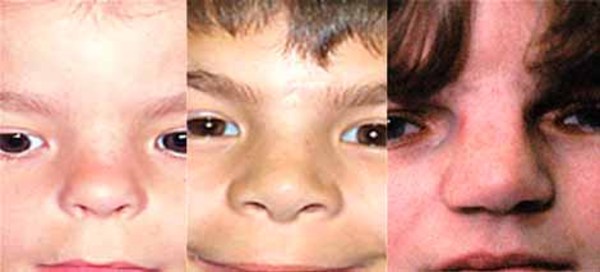
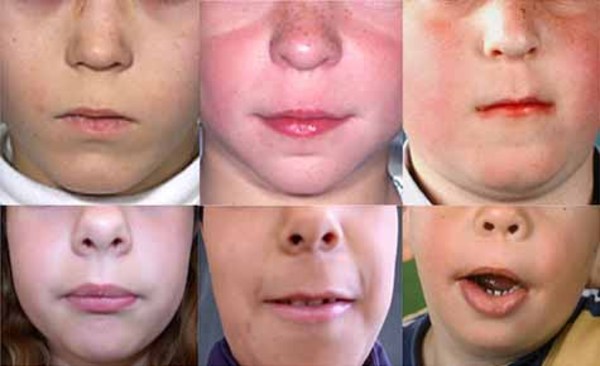

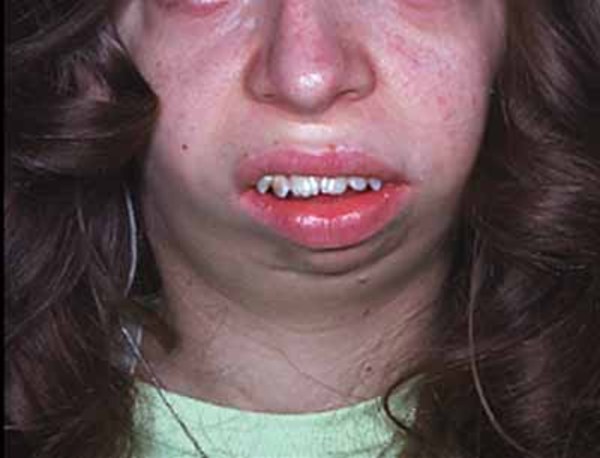
Other possible signs of this condition include dental problems, weak muscle tone (hypotonia), and hearing loss.
Frequency
The Maat-Kievit-Brunner type of Ohdo syndrome is a very rare condition, with only a few affected individuals reported in the medical literature.
Causes
The Maat-Kievit-Brunner type of Ohdo syndrome results from mutations in the MED12 gene. This gene provides instructions for making a protein that helps regulate gene activity; it is thought to play an essential role in development both before and after birth. The MED12 gene mutations that cause this condition alter the structure of the MED12 protein, impairing its ability to control gene activity. It is unclear how these changes lead to the particular cognitive and physical features of the Maat-Kievit-Brunner type of Ohdo syndrome.
Inheritance
This condition is inherited in an X-linked recessive pattern. The MED12 gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder.

Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. Females with only one altered copy of the gene in each cell are called carriers. They do not usually experience health problems related to the condition, but they can pass the mutation to their children. Sons who inherit the altered gene will have the condition, while daughters who inherit the altered gene will be carriers.
A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Blepharophimosis-mental retardation syndrome, Maat-Kievit-Brunner type
- BMRS, MKB type
- Ohdo syndrome, MKB type
- X-linked Ohdo syndrome
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Maat-Kievit A, Brunner HG, Maaswinkel-Mooij P. Two additional cases of the Ohdo blepharophimosis syndrome. Am J Med Genet. 1993 Nov 1;47(6):901-6. doi: 10.1002/ajmg.1320470618. Citation on PubMed
- Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, Leroy MA, Stevens R, Gillerot Y, Heron D, Heron B, Benzacken B, Lacombe D, Brunner H, Bitoun P. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006 Jun 15;140(12):1285-96. doi: 10.1002/ajmg.a.31270. Citation on PubMed
- Vulto-van Silfhout AT, de Vries BB, van Bon BW, Hoischen A, Ruiterkamp-Versteeg M, Gilissen C, Gao F, van Zwam M, Harteveld CL, van Essen AJ, Hamel BC, Kleefstra T, Willemsen MA, Yntema HG, van Bokhoven H, Brunner HG, Boyer TG, de Brouwer AP. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet. 2013 Mar 7;92(3):401-6. doi: 10.1016/j.ajhg.2013.01.007. Epub 2013 Feb 7. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.