

Description
Oculopharyngeal muscular dystrophy is a genetic condition characterized by muscle weakness that begins in adulthood, typically after age 40. The term "oculopharyngeal" refers to the eyes (oculo-) and a part of the throat called the pharynx (-pharyngeal). Affected individuals usually first experience weakness of the muscles in both eyelids that causes droopy eyelids (ptosis). Ptosis can worsen over time, causing the eyelid to impair vision, and in some cases, limit eye movement. Along with ptosis, affected individuals develop weakness of the throat muscles that causes difficulty swallowing (dysphagia). Dysphagia begins with dry food, but over time, liquids can also become difficult to swallow. Dysphagia can cause saliva to accumulate and a wet-sounding voice. Many people with oculopharyngeal muscular dystrophy also have weakness and wasting (atrophy) of the tongue. These problems with food intake may cause malnutrition, choking, or a bacterial lung infection called aspiration pneumonia.
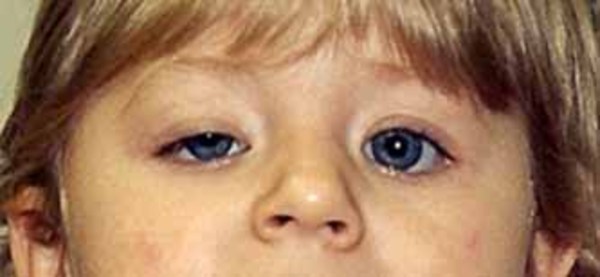
Individuals with oculopharyngeal muscular dystrophy frequently have weakness in the muscles near the center of the body (proximal muscles), particularly muscles in the shoulders, upper legs, and hips (limb-girdle muscles). The weakness slowly gets worse, and people may need the aid of a cane or a walker. Rarely, affected individuals need wheelchair assistance.
Rarely, individuals have a severe form of oculopharyngeal muscular dystrophy with muscle weakness that begins before age 45, and have trouble walking independently by age 60. These individuals often also have disturbances in nerve function (neuropathy), a gradual loss of intellectual functioning (cognitive decline), and psychiatric symptoms such as depression or strongly held false beliefs (delusions).
Frequency
In Europe, the prevalence of oculopharyngeal muscular dystrophy is estimated to be 1 in 100,000 people. This condition is much more common in the French-Canadian population of the Canadian province of Quebec, where it is estimated to affect 1 in 1,000 individuals. Oculopharyngeal muscular dystrophy is also seen more frequently in the Bukaran Jewish population of Israel, affecting 1 in 700 people.
Causes
Mutations in the PABPN1 gene cause oculopharyngeal muscular dystrophy. The PABPN1 gene provides instructions for making a protein that is found throughout the body. The PABPN1 protein plays an important role in processing molecules called messenger RNAs (mRNAs), which serve as genetic blueprints for making proteins. PABPN1 alters a region at the end of mRNA molecules that protects mRNA from being broken down. The PABPN1 protein also is involved in transporting mRNA within the cell.
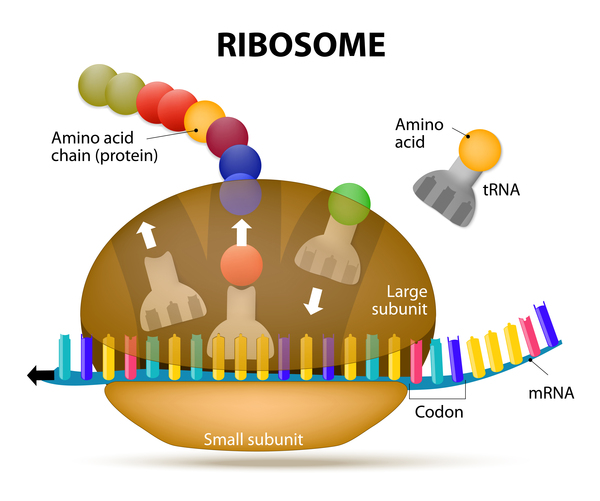
The PABPN1 protein contains an area where 10 copies of the protein building block (amino acid) alanine occur in a row. This stretch of alanines is known as a polyalanine tract. The role of the polyalanine tract in normal PABPN1 protein function is unknown. Mutations in the PABPN1 gene that cause oculopharyngeal muscular dystrophy result in a PABPN1 protein with an abnormally long (extended) polyalanine tract that includes between 11 and 18 alanines. Typically, affected individuals with shorter polyalanine tracts tend to have milder signs and symptoms that develop later in life compared to those with longer polyalanine tracts. The extra alanines cause the PABPN1 protein to form nonfunctional clumps within muscle cells. These clumps (called intranuclear inclusions) accumulate and are thought to impair the normal functioning of muscle cells, eventually causing cell death. The resulting loss of muscle cells over time most likely causes the muscle weakness seen in people with oculopharyngeal muscular dystrophy. In severe cases, it is likely that intranuclear inclusions affect nerve cells as well as muscle cells.

Inheritance
Most cases of oculopharyngeal muscular dystrophy are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. However, some individuals have mutations in both copies of the PABPN1 gene that lead to expanded polyalanine tracts. These individuals tend to have more severe signs and symptoms that develop earlier in life compared to individuals with a mutation in one copy of the gene.

In most cases, an affected person has one parent with the condition.
Other Names for This Condition
- Dystrophy, oculopharyngeal muscular
- Muscular dystrophy, oculopharyngeal
- Oculopharyngeal dystrophy
- OPMD
- Progressive muscular dystrophy, oculopharyngeal type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Blumen SC, Bouchard JP, Brais B, Carasso RL, Paleacu D, Drory VE, Chantal S, Blumen N, Braverman I. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology. 2009 Aug 25;73(8):596-601. doi: 10.1212/WNL.0b013e3181b388a3. Citation on PubMed
- Cruz-Aguilar M, Guerrero-de Ferran C, Tovilla-Canales JL, Nava-Castaneda A, Zenteno JC. Characterization of PABPN1 expansion mutations in a large cohort of Mexican patients with oculopharyngeal muscular dystrophy (OPMD). J Investig Med. 2017 Mar;65(3):705-708. doi: 10.1136/jim-2016-000184. Epub 2016 Dec 15. Citation on PubMed
- Richard P, Trollet C, Gidaro T, Demay L, Brochier G, Malfatti E, Tom FM, Fardeau M, Lafor P, Romero N, Martin-N ML, Sol G, Ferrer-Monasterio X, Saint-Guily JL, Eymard B. PABPN1 (GCN)11 as a Dominant Allele in Oculopharyngeal Muscular Dystrophy -Consequences in Clinical Diagnosis and Genetic Counselling. J Neuromuscul Dis. 2015 Jun 4;2(2):175-180. doi: 10.3233/JND-140060. Citation on PubMed or Free article on PubMed Central
- Richard P, Trollet C, Stojkovic T, de Becdelievre A, Perie S, Pouget J, Eymard B; Neurologists of French Neuromuscular Reference Centers CORNEMUS and FILNEMUS. Correlation between PABPN1 genotype and disease severity in oculopharyngeal muscular dystrophy. Neurology. 2017 Jan 24;88(4):359-365. doi: 10.1212/WNL.0000000000003554. Epub 2016 Dec 23. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.