

Description
Myotonic dystrophy is part of a group of inherited disorders called muscular dystrophies. It is the most common form of muscular dystrophy that begins in adulthood.
Myotonic dystrophy is characterized by progressive muscle wasting and weakness. People with this disorder often have prolonged muscle contractions (myotonia) and are not able to relax certain muscles after use. For example, a person may have difficulty releasing their grip on a doorknob or handle. Also, affected people may have slurred speech or temporary locking of their jaw.
Other signs and symptoms of myotonic dystrophy include clouding of the lens of the eye (cataracts) and abnormalities of the electrical signals that control the heartbeat (cardiac conduction defects). Some affected individuals develop a condition called diabetes mellitus, in which blood sugar (glucose) levels can become dangerously high. The features of myotonic dystrophy often develop during a person's twenties or thirties, although they can occur at any age. The severity of the condition varies widely among affected people, even among members of the same family.
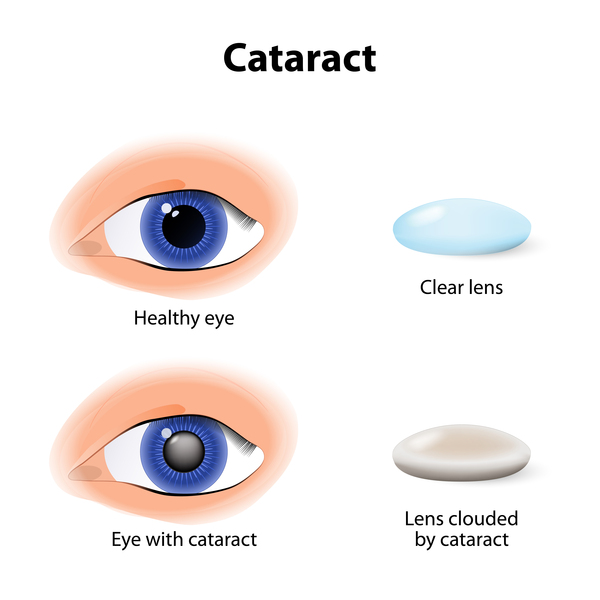
There are two major types of myotonic dystrophy: type 1 and type 2. Their signs and symptoms overlap, although type 2 tends to be milder than type 1. The muscle weakness associated with type 1 particularly affects muscles farthest from the center of the body (distal muscles), such as those of the lower legs, hands, neck, and face. Muscle weakness in type 2 primarily involves muscles close to the center of the body (proximal muscles), such as the those of the neck, shoulders, elbows, and hips. The two types of myotonic dystrophy are caused by mutations in different genes.
There are two variations of myotonic dystrophy type 1: the mild and congenital types. Mild myotonic dystrophy is apparent in mid to late adulthood. Affected individuals typically have mild myotonia and cataracts. Congenital myotonic dystrophy is often apparent at birth. Characteristic features include weak muscle tone (hypotonia), an inward- and upward-turning foot (clubfoot), breathing problems, delayed development, and intellectual disability. Some of these health problems can be life-threatening.

Frequency
Myotonic dystrophy affects at least 1 in 8,000 people worldwide. The prevalence of the two types of myotonic dystrophy varies among different geographic and ethnic populations. In most populations, type 1 appears to be more common than type 2. However, recent studies suggest that type 2 may be as common as type 1 among people in Germany and Finland.
Causes
Myotonic dystrophy type 1 is caused by mutations in the DMPK gene, while type 2 results from mutations in the CNBP gene. The protein produced from the DMPK gene likely plays a role in communication within cells. It appears to be important for the correct functioning of cells in the heart, brain, and skeletal muscles (which are used for movement). The protein produced from the CNBP gene is found primarily in the heart and in skeletal muscles, where it helps regulate the function of other genes.
Similar changes in the structure of the DMPK and CNBP genes cause myotonic dystrophy type 1 and type 2. In each case, a segment of DNA is abnormally repeated many times, forming an unstable region in the gene. The gene with the abnormal segment produces an unusually long messenger RNA, which is a molecular blueprint of the gene that guides the production of proteins. The unusually long messenger RNA forms clumps inside the cell that interfere with the production of many other proteins. These changes prevent muscle cells and cells in other tissues from functioning normally, which leads to the signs and symptoms of myotonic dystrophy. If these changes affect the DMPK gene, the result is myotonic dystrophy type 1, if the CNBP gene is affected, the result is myotonic dystrophy type 2.
Inheritance
Both types of myotonic dystrophy are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.

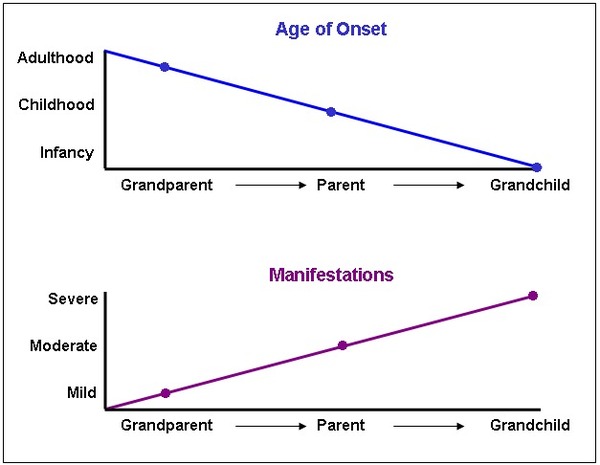
As myotonic dystrophy is passed from one generation to the next, the disorder generally begins earlier in life and signs and symptoms become more severe. This phenomenon is called anticipation. The evidence for anticipation appears only in myotonic dystrophy type 1. In this form of the disorder, anticipation is caused by an increase in the length of the unstable region in the DMPK gene. A longer unstable region in the CNBP gene does not appear to influence the age of onset of myotonic dystrophy type 2.
Other Names for This Condition
- Dystrophia myotonica
- Myotonia atrophica
- Myotonia dystrophica
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bird TD. Myotonic Dystrophy Type 1. 1999 Sep 17 [updated 2024 Nov 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1165/ Citation on PubMed
- Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003 Feb 25;60(4):657-64. doi: 10.1212/01.wnl.0000054481.84978.f9. Citation on PubMed
- Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science. 2004 Jan 16;303(5656):383-7. doi: 10.1126/science.1088679. Epub 2003 Dec 4. Citation on PubMed
- Kleefeld F, Erdmann H, Schoser B. Myotonic Dystrophy Type 2. 2006 Sep 21 [updated 2025 Sep 25]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1466/ Citation on PubMed
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001 Aug 3;293(5531):864-7. doi: 10.1126/science.1062125. Citation on PubMed
- Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review. Muscle Nerve. 2005 Jul;32(1):1-18. doi: 10.1002/mus.20301. Citation on PubMed
- Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 2015 Apr;1852(4):594-606. doi: 10.1016/j.bbadis.2014.05.019. Epub 2014 May 29. Citation on PubMed
- Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004 May;74(5):793-804. doi: 10.1086/383590. Epub 2004 Apr 2. Citation on PubMed or Free article on PubMed Central
- Thomas JD, Oliveira R, Sznajder LJ, Swanson MS. Myotonic Dystrophy and Developmental Regulation of RNA Processing. Compr Physiol. 2018 Mar 25;8(2):509-553. doi: 10.1002/cphy.c170002. Citation on PubMed
- Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, Kress W, Schoser B, Moxley R. 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006 Jun;16(6):403-13. doi: 10.1016/j.nmd.2006.03.010. Epub 2006 May 8. No abstract available. Citation on PubMed
- Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007 Oct;20(5):572-6. doi: 10.1097/WCO.0b013e3282ef6064. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.