

Description
Multiminicore disease is a disorder that primarily affects muscles used for movement (skeletal muscles). This condition causes muscle weakness and related health problems that range from mild to life-threatening.
Researchers have identified at least four forms of multiminicore disease, which can be distinguished by their characteristic signs and symptoms. The forms of multiminicore disease are the classic form, the progressive form with hand involvement, the antenatal form with arthrogryposis, and the ophthalmoplegic form.
The classic form accounts for about 75 percent of cases of multiminicore disease. This form causes muscle weakness beginning in infancy or early childhood. The muscles of the torso and neck (axial muscles) are most affected with arm and leg muscles less so. Muscle weakness causes affected infants to appear "floppy" (hypotonic) and they may have feeding problems early in life. Muscle weakness can delay the development of motor skills such as sitting, standing, and walking. In this form, the muscles of the ribcage and spine become stiff. In addition, the muscles needed for breathing are weak. This combination of muscle weakness and stiffness leads to severe or life-threatening respiratory problems. Almost all children with the classic form develop an abnormal curvature of the spine (scoliosis), which appears during childhood and steadily worsens over time.

The progressive form with hand involvement causes muscle weakness and looseness of the joints (joint laxity) in the arms and hands. Individuals with this form may experience muscle pain (myalgia) or extreme fatigue in response to physical activity (exercise intolerance). This form accounts for about 10 percent of cases of multiminicore disease.
The antenatal form with arthrogryposis is characterized by stiff, rigid joints throughout the body (arthrogryposis) and distinctive facial features. Weakness in the muscles needed for breathing can result in breathing problems for affected individuals. This form also accounts for about 10 percent of cases of multiminicore disease.
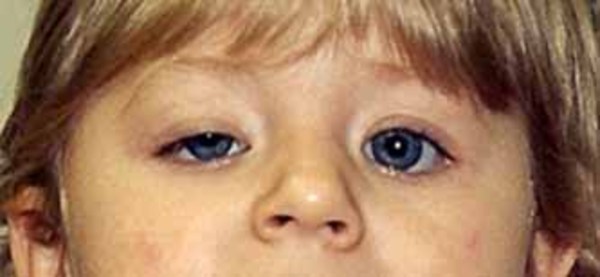
The ophthalmoplegic form of multiminicore disease is characterized by paralysis of the eye muscles (external ophthalmoplegia). This can lead to abnormal eye movements and droopy eyelids (ptosis). This form of the condition can also cause weakness in the muscles close to the center of the body (proximal muscles), such as those of the upper arms and legs. The ophthalmoplegic form accounts for 5 to 10 percent of cases of multiminicore disease.
Many people with multiminicore disease also have an increased risk of developing a severe reaction to certain drugs used during surgery and other invasive procedures. This reaction is called malignant hyperthermia. Malignant hyperthermia occurs in response to some anesthetic gases, which are used to block the sensation of pain, either given alone or in combination with a muscle relaxant that is used to temporarily paralyze a person during a surgical procedure. If given these drugs, people at risk of malignant hyperthermia may experience a rapid increase in heart rate (tachycardia) and body temperature (hyperthermia), abnormally fast breathing (tachypnea), muscle rigidity, breakdown of muscle fibers (rhabdomyolysis), and increased acid levels in the blood and other tissues (acidosis). The complications of malignant hyperthermia can be life-threatening unless they are treated promptly.
Multiminicore disease gets its name from small, disorganized areas called minicores, which are found in skeletal muscle cells of many affected individuals. These abnormal regions can only been seen when muscle tissue is viewed under a microscope. Minicores are often present in cells with few or no mitochondria, which are the energy-producing centers within cells. Although the presence of minicores can help doctors diagnose multiminicore disease, it is unclear how they are related to muscle weakness and the other features of this condition.

Frequency
Multiminicore disease is thought to be a rare disorder, although its incidence is unknown.
Causes
Variants (also known as mutations) in the SELENON and RYR1 genes have been found to cause about half of all cases of multiminicore disease.
About 30 percent of cases of multiminicore disease, primarily the classic form, are caused by variants in the SELENON gene. This gene provides instructions for making a protein called selenoprotein N. This protein is highly active in many tissues before birth and may be involved in the formation of muscle tissue (myogenesis). The protein may also be important for normal muscle function after birth, although it is active at much lower levels in adult tissues. This protein is thought to play a role in maintaining an appropriate balance of calcium (calcium homeostasis) in cells. Calcium plays an important role in muscle movement. It is unclear, however, how variants in the SELENON gene lead to muscle weakness and the other features of multiminicore disease.
An estimated 20 percent of multiminicore disease, primarily the non-classic forms, are caused by variants in the RYR1 gene. The RYR1 gene provides instructions for making a protein called ryanodine receptor 1. This protein plays an essential role in skeletal muscles. For the body to move normally, these muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of charged atoms (ions) into muscle cells. The ryanodine receptor 1 protein forms a channel that releases calcium ions stored within muscle cells. The resulting increase in calcium ion concentration inside muscle cells stimulates muscle fibers to contract, allowing the body to move.
Variants in the RYR1 gene change the structure and function of the ryanodine receptor 1 protein and the calcium channel that it forms. The abnormal calcium channel alters the normal flow of stored calcium ions within muscle cells. A disruption in calcium ion transport prevents muscles from contracting normally, leading to the muscle weakness characteristic of multiminicore disease. RYR1 gene variants are also associated with an increased risk of malignant hyperthermia.
It is likely that individuals with multiminicore disease who do not have a known variant in either of these two genes have variants in other genes that underlie the condition.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Minicore disease
- Minicore myopathy
- MmD
- Multi-core congenital myopathy
- Multi-core disease
- Multi-minicore disease
- Multicore disease
- Multicore myopathy
- Multiminicore myopathy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ferreiro A, Estournet B, Chateau D, Romero NB, Laroche C, Odent S, Toutain A, Cabello A, Fontan D, dos Santos HG, Haenggeli CA, Bertini E, Urtizberea JA, Guicheney P, Fardeau M. Multi-minicore disease--searching for boundaries: phenotype analysis of 38 cases. Ann Neurol. 2000 Nov;48(5):745-57. Citation on PubMed
- Ferreiro A, Quijano-Roy S, Pichereau C, Moghadaszadeh B, Goemans N, Bonnemann C, Jungbluth H, Straub V, Villanova M, Leroy JP, Romero NB, Martin JJ, Muntoni F, Voit T, Estournet B, Richard P, Fardeau M, Guicheney P. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002 Oct;71(4):739-49. doi: 10.1086/342719. Epub 2002 Aug 21. Citation on PubMed or Free article on PubMed Central
- Fusto A, Moyle LA, Gilbert PM, Pegoraro E. Cored in the act: the use of models to understand core myopathies. Dis Model Mech. 2019 Dec 19;12(12):dmm041368. doi: 10.1242/dmm.041368. Citation on PubMed or Free article on PubMed Central
- Jungbluth H, Beggs A, Bonnemann C, Bushby K, Ceuterick-de Groote C, Estournet-Mathiaud B, Goemans N, Guicheney P, Lescure A, Lunardi J, Muntoni F, Quinlivan R, Sewry C, Straub V, Treves S, Ferreiro A. 111th ENMC International Workshop on Multi-minicore Disease. 2nd International MmD Workshop, 9-11 November 2002, Naarden, The Netherlands. Neuromuscul Disord. 2004 Nov;14(11):754-66. doi: 10.1016/j.nmd.2004.07.007. No abstract available. Citation on PubMed
- Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry C, Phadke R, Gautel M, Muntoni F. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol. 2018 Mar;14(3):151-167. doi: 10.1038/nrneurol.2017.191. Epub 2018 Feb 2. Citation on PubMed
- Jungbluth H. Multi-minicore Disease. Orphanet J Rare Dis. 2007 Jul 13;2:31. doi: 10.1186/1750-1172-2-31. Citation on PubMed or Free article on PubMed Central
- Lawal TA, Todd JJ, Meilleur KG. Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches. Neurotherapeutics. 2018 Oct;15(4):885-899. doi: 10.1007/s13311-018-00677-1. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.