

Description
Multicentric osteolysis, nodulosis, and arthropathy (MONA) describes a rare inherited disease characterized by a loss of bone tissue (osteolysis), particularly in the hands and feet. MONA includes a condition formerly called nodulosis-arthropathy-osteolysis (NAO) syndrome. It may also include a similar disorder called Torg syndrome, although it is unknown whether Torg syndrome is actually part of MONA or a separate disorder caused by a mutation in a different gene.
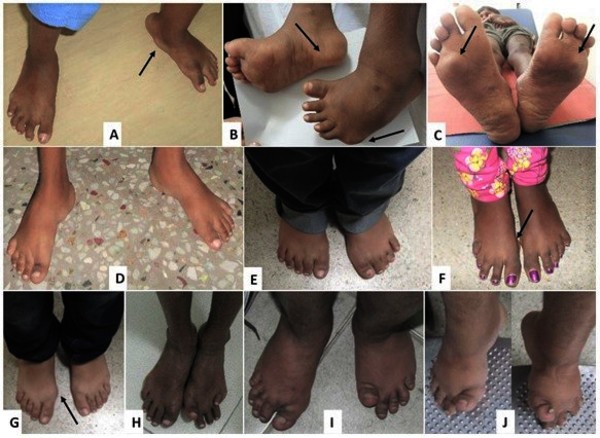
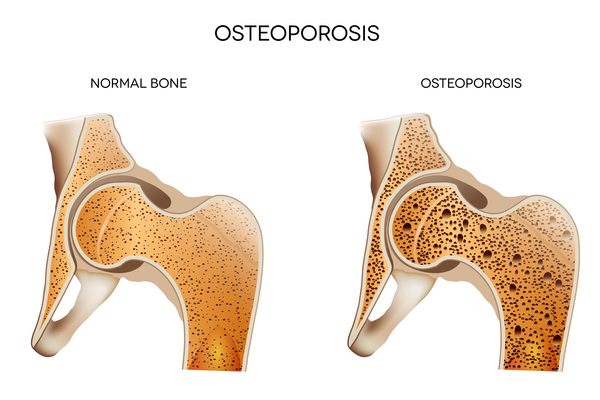
In most cases of MONA, bone loss begins in the hands and feet, causing pain and limiting movement. Bone abnormalities can later spread to other areas of the body, with joint problems (arthropathy) occurring in the elbows, shoulders, knees, hips, and spine. Most people with MONA develop low bone mineral density (osteopenia) and thinning of the bones (osteoporosis) throughout the skeleton. These abnormalities make bones brittle and more prone to fracture. The bone abnormalities also lead to short stature.
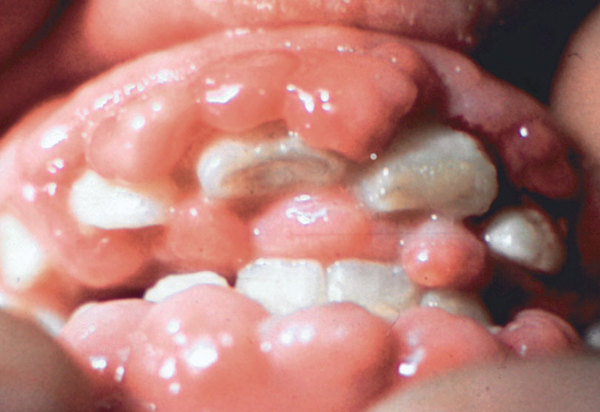
Many affected individuals develop subcutaneous nodules, which are firm lumps of noncancerous tissue underneath the skin, especially on the soles of the feet. Some affected individuals also have skin abnormalities including patches of dark, thick, and leathery skin. Other features of MONA can include clouding of the clear front covering of the eye (corneal opacity), excess hair growth (hypertrichosis), overgrowth of the gums, heart abnormalities, and distinctive facial features that are described as "coarse."

Frequency
MONA is rare; its prevalence is unknown. This condition has been reported in multiple populations worldwide.
Causes
MONA results from mutations in the MMP2 gene. This gene provides instructions for making an enzyme called matrix metallopeptidase 2, whose primary function is to cut (cleave) a protein called type IV collagen. Type IV collagen is a major structural component of basement membranes, which are thin, sheet-like structures that separate and support cells in many tissues. The activity of matrix metallopeptidase 2 appears to be important for a variety of body functions, including bone remodeling, which is a normal process in which old bone is broken down and new bone is created to replace it.
The MMP2 gene mutations that cause MONA completely eliminate the activity of the matrix metallopeptidase 2 enzyme, preventing the normal cleavage of type IV collagen. It is unclear how a loss of enzyme activity leads to the specific features of MONA. Researchers suspect that it somehow disrupts the balance of new bone creation and the breakdown of existing bone during bone remodeling, resulting in a progressive loss of bone tissue. How a shortage of matrix metallopeptidase 2 leads to the other features of MONA, such as subcutaneous nodules and skin abnormalities, is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Al-Aqeel Sewairi syndrome
- Hereditary multicentric osteolysis
- MONA
- NAO syndrome
- Nodulosis-arthropathy-osteolysis syndrome
- Torg syndrome
- Torg-Winchester syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Al Aqeel A, Al Sewairi W, Edress B, Gorlin RJ, Desnick RJ, Martignetti JA. Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am J Med Genet. 2000 Jul 3;93(1):11-8. doi: 10.1002/1096-8628(20000703)93:13.0.co;2-3. Citation on PubMed
- Al-Mayouf SM, Majeed M, Hugosson C, Bahabri S. New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am J Med Genet. 2000 Jul 3;93(1):5-10. doi: 10.1002/1096-8628(20000703)93:13.0.co;2-y. Citation on PubMed
- Castberg FC, Kjaergaard S, Mosig RA, Lobl M, Martignetti C, Martignetti JA, Myrup C, Zak M. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur J Pediatr. 2013 Dec;172(12):1657-63. doi: 10.1007/s00431-013-2102-8. Epub 2013 Jul 31. Citation on PubMed
- Evans BR, Mosig RA, Lobl M, Martignetti CR, Camacho C, Grum-Tokars V, Glucksman MJ, Martignetti JA. Mutation of membrane type-1 metalloproteinase, MT1-MMP, causes the multicentric osteolysis and arthritis disease Winchester syndrome. Am J Hum Genet. 2012 Sep 7;91(3):572-6. doi: 10.1016/j.ajhg.2012.07.022. Epub 2012 Aug 23. Citation on PubMed or Free article on PubMed Central
- Martignetti JA, Aqeel AA, Sewairi WA, Boumah CE, Kambouris M, Mayouf SA, Sheth KV, Eid WA, Dowling O, Harris J, Glucksman MJ, Bahabri S, Meyer BF, Desnick RJ. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001 Jul;28(3):261-5. doi: 10.1038/90100. Citation on PubMed
- Rouzier C, Vanatka R, Bannwarth S, Philip N, Coussement A, Paquis-Flucklinger V, Lambert JC. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin Genet. 2006 Mar;69(3):271-6. doi: 10.1111/j.1399-0004.2006.00584.x. Citation on PubMed
- Tuysuz B, Mosig R, Altun G, Sancak S, Glucksman MJ, Martignetti JA. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 2009 May;17(5):565-72. doi: 10.1038/ejhg.2008.204. Epub 2008 Nov 5. Citation on PubMed or Free article on PubMed Central
- Zankl A, Bonafe L, Calcaterra V, Di Rocco M, Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet. 2005 Mar;67(3):261-6. doi: 10.1111/j.1399-0004.2004.00402.x. Citation on PubMed
- Zankl A, Pachman L, Poznanski A, Bonafe L, Wang F, Shusterman Y, Fishman DA, Superti-Furga A. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J Bone Miner Res. 2007 Feb;22(2):329-33. doi: 10.1359/jbmr.061013. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.