

Description
Mucopolysaccharidosis type VI (MPS VI), also known as Maroteaux-Lamy syndrome, is a progressive condition that causes many tissues and organs to enlarge, become inflamed or scarred, and eventually waste away (atrophy). Skeletal abnormalities are also common in this condition. The rate at which symptoms worsen varies among affected individuals.
People with MPS VI generally do not display any features of the condition at birth. They often begin to show signs and symptoms of MPS VI during early childhood. The features of MPS VI affect many bodily systems, including skeletal, cardiac, and respiratory.
MPS VI causes various skeletal abnormalities, including a large head (macrocephaly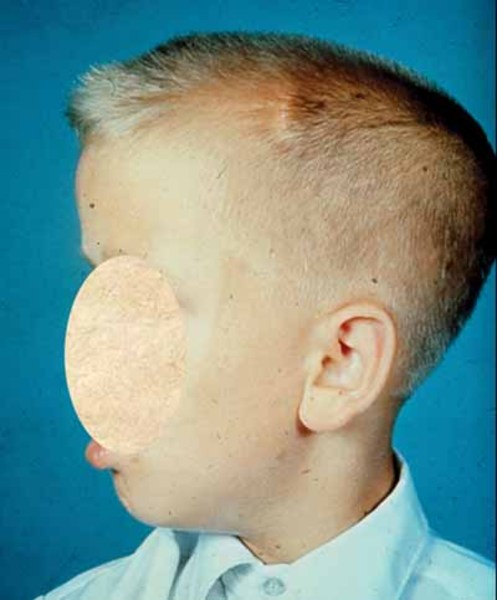 ) with a buildup of fluid in the brain (hydrocephalus), distinctive-looking facial features
) with a buildup of fluid in the brain (hydrocephalus), distinctive-looking facial features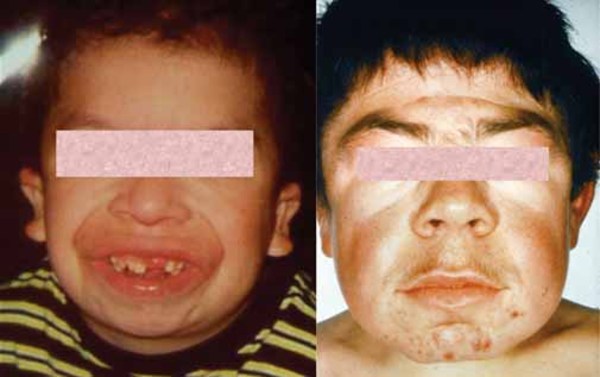 that are described as "coarse," and a large tongue (macroglossia
that are described as "coarse," and a large tongue (macroglossia ). Other skeletal features include short stature, joint deformities (contractures) that affect mobility, and dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Carpal tunnel syndrome develops in many children with MPS VI and is characterized by numbness, tingling, and weakness in the hands and fingers. People with MPS VI may develop a narrowing of the spinal canal (spinal stenosis
). Other skeletal features include short stature, joint deformities (contractures) that affect mobility, and dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Carpal tunnel syndrome develops in many children with MPS VI and is characterized by numbness, tingling, and weakness in the hands and fingers. People with MPS VI may develop a narrowing of the spinal canal (spinal stenosis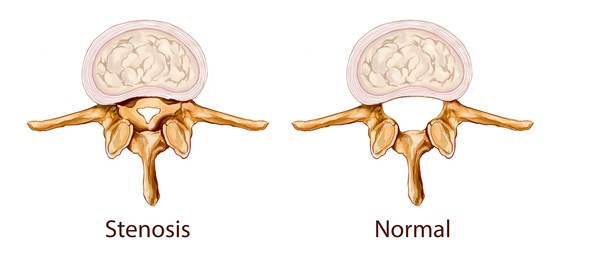 ) in the neck, which can compress and damage the spinal cord.
) in the neck, which can compress and damage the spinal cord.
Cardiac problems in people with MPS VI typically includes heart valve abnormalities. Respiratory abnormalities in this condition may involve the airway becoming narrow, which leads to frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea).
Other features of MPS VI include an enlarged liver and spleen (hepatosplenomegaly), and a soft out-pouching around the belly-button (umbilical hernia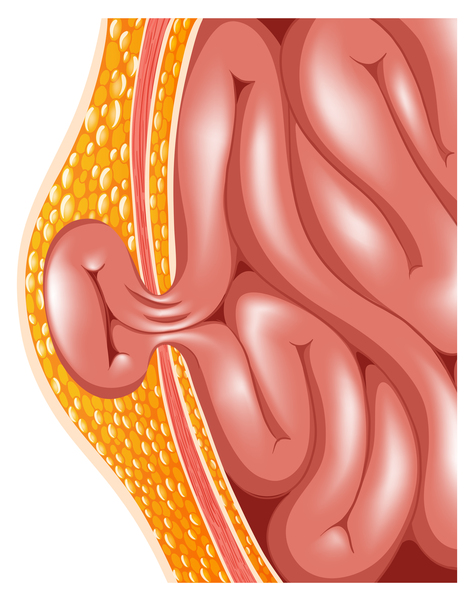 ) or lower abdomen (inguinal hernia). The clear covering of the eye (cornea
) or lower abdomen (inguinal hernia). The clear covering of the eye (cornea ) typically becomes cloudy, which can cause significant vision loss. People with MPS VI may also have recurrent ear infections and hearing loss. Unlike other types of mucopolysaccharidosis, MPS VI does not affect intelligence.
) typically becomes cloudy, which can cause significant vision loss. People with MPS VI may also have recurrent ear infections and hearing loss. Unlike other types of mucopolysaccharidosis, MPS VI does not affect intelligence.
The life expectancy of individuals with MPS VI depends on the severity of symptoms. Without treatment, severely affected individuals may survive only until late childhood or adolescence. Those with milder forms of the disorder usually live into adulthood, although their life expectancy may be reduced. Heart disease and airway obstruction are major causes of death in people with MPS VI.
Frequency
The incidence of MPS VI is unknown, although it is estimated to occur in 1 in 250,000 to 600,000 newborns.
Causes
Mutations in the ARSB gene cause MPS VI. The ARSB gene provides instructions for producing an enzyme called arylsulfatase B (also known as N-acetylgalactosamine-4-sulfatase), which is involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). GAGs were originally called mucopolysaccharides, which is where this condition gets its name.
Mutations in the ARSB gene reduce or completely eliminate the function of arylsulfatase B. The lack of arylsulfatase B activity leads to the accumulation of GAGs within cells, specifically inside the lysosomes . Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as MPS VI that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. The accumulation of GAGs within lysosomes increases the size of the cells, which is why many tissues and organs are enlarged in this disorder. Researchers believe that the buildup of GAGs are toxic to cells and may also interfere with the functions of other proteins inside lysosomes, triggering inflammation and cell death. The loss of cells leads to atrophy of tissues and organs over time in MPS VI.
. Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as MPS VI that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. The accumulation of GAGs within lysosomes increases the size of the cells, which is why many tissues and organs are enlarged in this disorder. Researchers believe that the buildup of GAGs are toxic to cells and may also interfere with the functions of other proteins inside lysosomes, triggering inflammation and cell death. The loss of cells leads to atrophy of tissues and organs over time in MPS VI.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Arylsulfatase B deficiency
- Maroteaux-Lamy syndrome
- MPS VI
- MPS6
- Mucopolysaccharidosis 6
- Mucopolysaccharidosis VI
- Polydystrophic dwarfism
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Azevedo AC, Schwartz IV, Kalakun L, Brustolin S, Burin MG, Beheregaray AP, Leistner S, Giugliani C, Rosa M, Barrios P, Marinho D, Esteves P, Valadares E, Boy R, Horovitz D, Mabe P, da Silva LC, de Souza IC, Ribeiro M, Martins AM, Palhares D, Kim CA, Giugliani R. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet. 2004 Sep;66(3):208-13. doi: 10.1111/j.1399-0004.2004.00277.x. Citation on PubMed
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschutter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28(6):1011-7. doi: 10.1007/s10545-005-0112-z. Citation on PubMed
- Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008 Jan 18;10:e1. doi: 10.1017/S1462399408000550. Citation on PubMed
- Garrido E, Chabas A, Coll MJ, Blanco M, Dominguez C, Grinberg D, Vilageliu L, Cormand B. Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Mol Genet Metab. 2007 Sep-Oct;92(1-2):122-30. doi: 10.1016/j.ymgme.2007.06.002. Epub 2007 Jul 20. Citation on PubMed
- Garrido E, Cormand B, Hopwood JJ, Chabas A, Grinberg D, Vilageliu L. Maroteaux-Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol Genet Metab. 2008 Jul;94(3):305-12. doi: 10.1016/j.ymgme.2008.02.012. Epub 2008 Apr 10. Citation on PubMed
- Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007 Aug;120(2):405-18. doi: 10.1542/peds.2006-2184. Citation on PubMed
- Karageorgos L, Brooks DA, Pollard A, Melville EL, Hein LK, Clements PR, Ketteridge D, Swiedler SJ, Beck M, Giugliani R, Harmatz P, Wraith JE, Guffon N, Leao Teles E, Sa Miranda MC, Hopwood JJ. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007 Sep;28(9):897-903. doi: 10.1002/humu.20534. Citation on PubMed
- Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, Chao MC, Chiu PC, Lin SJ, Tsai LP, Hwu WL, Lin JL. Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A. 2009 May;149A(5):960-4. doi: 10.1002/ajmg.a.32781. Citation on PubMed
- Litjens T, Hopwood JJ. Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat. 2001 Oct;18(4):282-95. doi: 10.1002/humu.1190. Citation on PubMed
- Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003 Dec 15;123A(3):310-3. doi: 10.1002/ajmg.a.20314. Citation on PubMed
- Tessitore A, Pirozzi M, Auricchio A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics. 2009 Jun 16;2(1):4. doi: 10.1186/1755-8417-2-4. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.