

Description
Mucopolysaccharidosis type III (MPS III), also known as Sanfilippo syndrome, is a disorder that primarily affects the brain and spinal cord (central nervous system). It is characterized by deterioration of neurological function (neurodegeneration), resulting in many of the features of the condition. Other body systems can also be involved, although the physical features are usually mild in the early stages.
People with MPS III generally do not display any features of the condition at birth, but they begin to show signs and symptoms of the disorder during early childhood . Early signs and symptoms of MPS III can include frequent ear and throat infections or bowel problems, though most common are mild developmental delay or delayed speech. Behavioral problems often worsen with affected children becoming restless, hyperactive, destructive, anxious, impulsive, fearless, or aggressive. Some affected children display features of autism spectrum disorder, which is a condition characterized by difficulty with social interactions and communication. Children with MPS III may have an increased tendency to chew on objects or put things in their mouth (be hyperoral). Sleep disturbances are also very common in children with MPS III. This condition causes progressive intellectual disability and the loss of previously acquired skills (developmental regression or dementia). In later stages of the disorder, people with MPS III may develop seizures, loss of mobility, and movement disorders.
. Early signs and symptoms of MPS III can include frequent ear and throat infections or bowel problems, though most common are mild developmental delay or delayed speech. Behavioral problems often worsen with affected children becoming restless, hyperactive, destructive, anxious, impulsive, fearless, or aggressive. Some affected children display features of autism spectrum disorder, which is a condition characterized by difficulty with social interactions and communication. Children with MPS III may have an increased tendency to chew on objects or put things in their mouth (be hyperoral). Sleep disturbances are also very common in children with MPS III. This condition causes progressive intellectual disability and the loss of previously acquired skills (developmental regression or dementia). In later stages of the disorder, people with MPS III may develop seizures, loss of mobility, and movement disorders.
The physical features of MPS III are less pronounced than those of other types of mucopolysaccharidosis. Individuals with MPS III typically have mildly "coarse" facial features , a prominent forehead, a large head (macrocephaly
, a prominent forehead, a large head (macrocephaly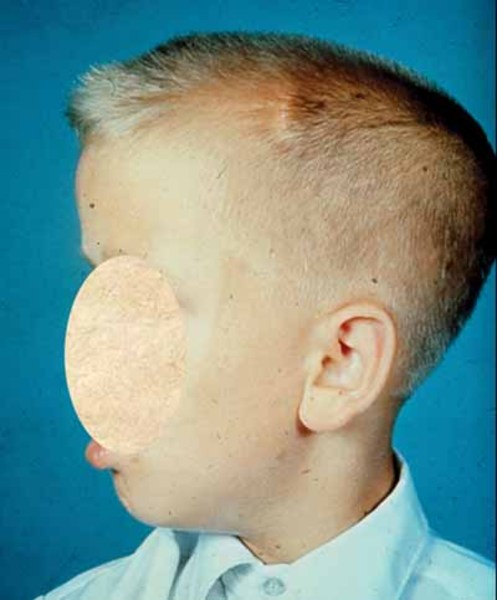 ), and thick hair and eyebrows. Some people with MPS III have short stature, joint stiffness, or mild dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray.
), and thick hair and eyebrows. Some people with MPS III have short stature, joint stiffness, or mild dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray.
People with MPS III often have a slightly enlarged liver (mild hepatomegaly) or spleen (mild splenomegaly), and a soft out-pouching around the belly-button (umbilical hernia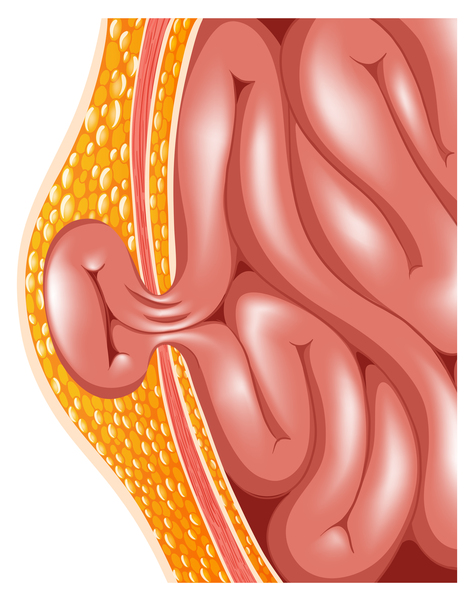 ) or lower abdomen (inguinal hernia). Cardiac abnormalities may also occur in this condition, including weakening of the heart muscle (cardiomyopathy), disruption of the heart's normal rhythm (arrhythmia), or problems with the heart's valves. Affected individuals often experience chronic diarrhea and recurrent upper respiratory and ear infections. People with MPS III
) or lower abdomen (inguinal hernia). Cardiac abnormalities may also occur in this condition, including weakening of the heart muscle (cardiomyopathy), disruption of the heart's normal rhythm (arrhythmia), or problems with the heart's valves. Affected individuals often experience chronic diarrhea and recurrent upper respiratory and ear infections. People with MPS III may also have hearing loss and vision problems.
may also have hearing loss and vision problems.
MPS III is divided into types IIIA, IIIB, IIIC, and IIID, which are distinguished by their genetic cause. The different types of MPS III have similar signs and symptoms, although the features of MPS IIIA typically appear earlier in life and progress more rapidly. People with MPS III usually live into adolescence or early to mid-adulthood.
Frequency
MPS III is the most common form of mucopolysaccharidosis; the estimated incidence of all four types combined is 1 in 70,000 newborns. MPS IIIA and MPS IIIB are much more common than MPS IIIC and MPS IIID.
Causes
Variants (also called mutations) in the GNS, HGSNAT, NAGLU, and SGSH genes cause MPS III. These genes provide instructions for making enzymes involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). GAGs were originally called mucopolysaccharides, which is where this condition gets its name. The GNS, HGSNAT, NAGLU, and SGSH enzymes are involved in the step-wise breakdown of a subset of GAGs called heparan sulfate.
MPS IIIA is caused by variants in the SGSH gene, and MPS IIIB is caused by NAGLU gene variants. Variants in the HGSNAT gene result in MPS IIIC, and GNS gene mutations cause MPS IIID. Variants in these genes reduce or eliminate enzyme function. A lack of any one of these enzymes disrupts the breakdown of heparan sulfate. As a result, partially broken down heparan sulfate accumulates within cells, specifically inside the lysosomes . Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as MPS III that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. Researchers believe that the accumulation of GAGs interferes with the functions of other proteins inside the lysosomes and disrupts the normal functions of cells. It is unknown why the buildup of heparan sulfate mostly affects the central nervous system in MPS III.
. Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as MPS III that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. Researchers believe that the accumulation of GAGs interferes with the functions of other proteins inside the lysosomes and disrupts the normal functions of cells. It is unknown why the buildup of heparan sulfate mostly affects the central nervous system in MPS III.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- MPS III
- Mucopolysaccharidosis III
- Sanfilippo syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bodamer OA, Giugliani R, Wood T. The laboratory diagnosis of mucopolysaccharidosis III (Sanfilippo syndrome): A changing landscape. Mol Genet Metab. 2014 Sep-Oct;113(1-2):34-41. doi: 10.1016/j.ymgme.2014.07.013. Epub 2014 Jul 16. Citation on PubMed
- Gilkes JA, Heldermon CD. Mucopolysaccharidosis III (Sanfilippo Syndrome)- disease presentation and experimental therapies. Pediatr Endocrinol Rev. 2014 Sep;12 Suppl 1:133-40. Citation on PubMed
- Malm G, Mansson JE. Mucopolysaccharidosis type III (Sanfilippo disease) in Sweden: clinical presentation of 22 children diagnosed during a 30-year period. Acta Paediatr. 2010 Aug;99(8):1253-7. doi: 10.1111/j.1651-2227.2010.01800.x. Epub 2010 Mar 14. Citation on PubMed
- Meyer A, Kossow K, Gal A, Muhlhausen C, Ullrich K, Braulke T, Muschol N. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics. 2007 Nov;120(5):e1255-61. doi: 10.1542/peds.2007-0282. Epub 2007 Oct 15. Citation on PubMed
- Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, Wevers RA, Poorthuis BJ, Pshezhetsky AV, Wijburg FA. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011. Epub 2007 Nov 19. Citation on PubMed
- Valstar MJ, Bertoli-Avella AM, Wessels MW, Ruijter GJ, de Graaf B, Olmer R, Elfferich P, Neijs S, Kariminejad R, Suheyl Ezgu F, Tokatli A, Czartoryska B, Bosschaart AN, van den Bos-Terpstra F, Puissant H, Burger F, Omran H, Eckert D, Filocamo M, Simeonov E, Willems PJ, Wevers RA, Niermeijer MF, Halley DJ, Poorthuis BJ, van Diggelen OP. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234. Citation on PubMed
- Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008 Apr;31(2):240-52. doi: 10.1007/s10545-008-0838-5. Epub 2008 Apr 4. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.