

Description
Melnick-Needles syndrome is a disorder involving abnormalities in skeletal development and other health problems. It is a member of a group of related conditions called otopalatodigital spectrum disorders, which also includes otopalatodigital syndrome type 1, otopalatodigital syndrome type 2, frontometaphyseal dysplasia, and terminal osseous dysplasia. In general, these disorders involve hearing loss caused by malformations in the tiny bones in the ears (ossicles ), problems in the development of the roof of the mouth (palate), and skeletal abnormalities involving the fingers and/or toes (digits).
), problems in the development of the roof of the mouth (palate), and skeletal abnormalities involving the fingers and/or toes (digits).
Melnick-Needles syndrome is usually the most severe of the otopalatodigital spectrum disorders. People with this condition are usually of short stature, have an abnormal curvature of the spine (scoliosis ), partial dislocation (subluxation) of certain joints, and unusually long fingers
), partial dislocation (subluxation) of certain joints, and unusually long fingers and toes. They may have bowed limbs; underdeveloped, irregular ribs that can cause problems with breathing; and other abnormal or absent bones.
and toes. They may have bowed limbs; underdeveloped, irregular ribs that can cause problems with breathing; and other abnormal or absent bones.
Characteristic facial features may include bulging eyes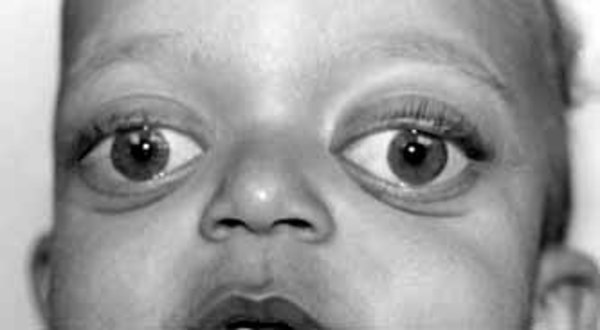 with prominent brow ridges, excess hair growth on the forehead, round cheeks, a very small lower jaw and chin (micrognathia
with prominent brow ridges, excess hair growth on the forehead, round cheeks, a very small lower jaw and chin (micrognathia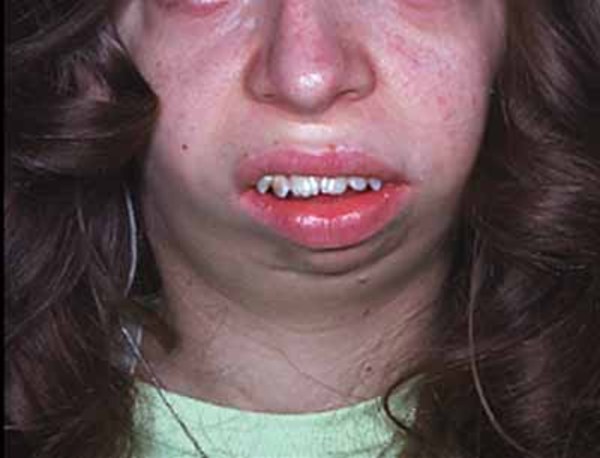 ), and misaligned teeth. One side of the face may appear noticeably different from the other (facial asymmetry). Some individuals with this disorder have hearing loss.
), and misaligned teeth. One side of the face may appear noticeably different from the other (facial asymmetry). Some individuals with this disorder have hearing loss.
In addition to skeletal abnormalities, individuals with Melnick-Needles syndrome may have obstruction of the ducts between the kidneys and bladder (ureters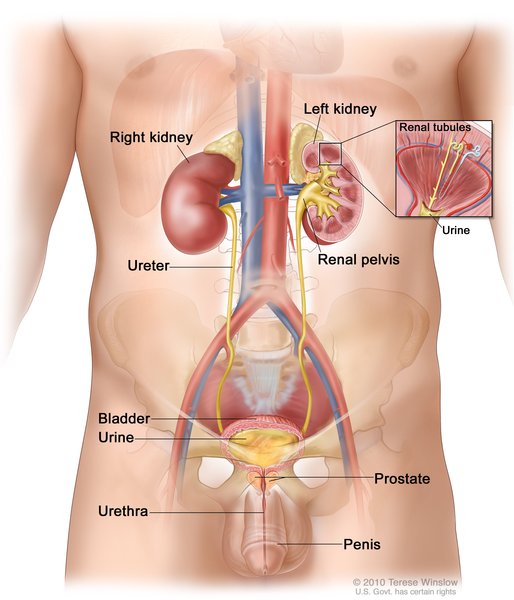 ) or heart defects.
) or heart defects.
Males with Melnick-Needles syndrome generally have much more severe signs and symptoms than do females, and in almost all cases die before or soon after birth.
Frequency
Melnick-Needles syndrome is a rare disorder; fewer than 100 cases have been reported worldwide.
Causes
Variants (also called mutations) in the FLNA gene cause Melnick-Needles syndrome.
The FLNA gene provides instructions for producing the protein filamin A, which helps build the network of protein filaments (cytoskeleton ) that gives structure to cells and allows them to change shape and move. Filamin A binds to another protein called actin, and helps the actin to form the branching network of filaments that make up the cytoskeleton. Filamin A also links actin to many other proteins to perform various functions within the cell.
) that gives structure to cells and allows them to change shape and move. Filamin A binds to another protein called actin, and helps the actin to form the branching network of filaments that make up the cytoskeleton. Filamin A also links actin to many other proteins to perform various functions within the cell.
A small number of variants in the FLNA gene have been identified in people with Melnick-Needles syndrome. These variants are described as "gain-of-function" because they appear to enhance the activity of the filamin A protein or give the protein a new, atypical function. Researchers believe that the variants may change the way the filamin A protein helps regulate processes involved in skeletal development, but it is not known how changes in the protein relate to the specific signs and symptoms of Melnick-Needles syndrome.
Inheritance
This condition is inherited in an X-linked dominant pattern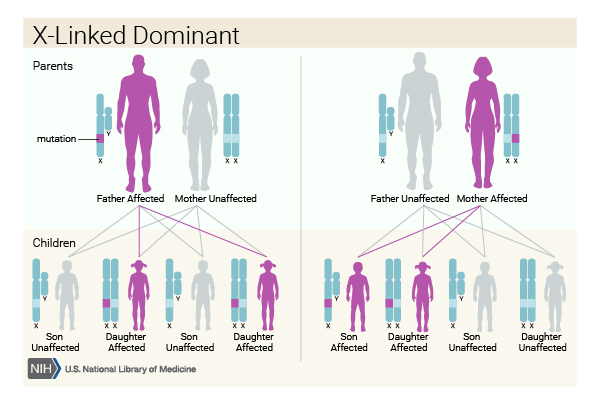 . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Melnick-Needles osteodysplasty
- MNS
- Osteodysplasty of Melnick and Needles
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Albano LMJ, Bertola DR, Barba MF, Valente M, Robertson SP, Kim CA. Phenotypic overlap in Melnick-Needles, serpentine fibula-polycystic kidney and Hajdu-Cheney syndromes: a clinical and molecular study in three patients. Clin Dysmorphol. 2007 Jan;16(1):27-33. doi: 10.1097/01.mcd.0000228418.74413.52. Citation on PubMed
- Kristiansen M, Knudsen GP, Soyland A, Westvik J, Orstavik KH. Phenotypic variation in Melnick-Needles syndrome is not reflected in X inactivation patterns from blood or buccal smear. Am J Med Genet. 2002 Mar 1;108(2):120-7. doi: 10.1002/ajmg.10245. Citation on PubMed
- Robertson S, Wade E. FLNA-Related Otopalatodigital Spectrum Disorders. 2005 Nov 30 [updated 2025 Jun 26]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1393/ Citation on PubMed
- Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, Orstavik KH, Quarrell OW, Schwartz CE, Shears DJ, Suri M, Kendrick-Jones J, Wilkie AO; OPD-spectrum Disorders Clinical Collaborative Group. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. 2003 Apr;33(4):487-91. doi: 10.1038/ng1119. Epub 2003 Mar 3. Citation on PubMed
- Robertson SP. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. Eur J Hum Genet. 2007 Jan;15(1):3-9. doi: 10.1038/sj.ejhg.5201654. Epub 2006 Aug 23. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.