

Description
MECP2 duplication syndrome is a condition that occurs almost exclusively in males and is characterized by moderate to severe intellectual disability. Most people with this condition also have weak muscle tone in infancy, feeding difficulties, poor or absent speech, or muscle stiffness (rigidity). Individuals with MECP2 duplication syndrome have delayed development of motor skills such as sitting and walking. About half of individuals have seizures, often of the tonic-clonic type. This type of seizure involves a loss of consciousness, muscle rigidity, and convulsions and may not respond to medication. Some affected individuals experience the loss of previously acquired skills (developmental regression). Approximately half of individuals learn to walk, and about one-third of people with this condition require assistance when walking. Many individuals with MECP2 duplication syndrome have recurrent respiratory tract infections. These respiratory infections are a major cause of death in affected individuals, with only half surviving past age 25.
Frequency
The prevalence of MECP2 duplication syndrome is unknown; more than 200 affected individuals have been described in the scientific literature. It is estimated that this condition is responsible for 1 to 2 percent of all cases of intellectual disability caused by changes in the X chromosome.
Causes
MECP2 duplication syndrome is caused by a genetic change in which there is an extra copy of the MECP2 gene in each cell. This extra copy of the MECP2 gene is caused by a duplication of genetic material on the long (q) arm of the X chromosome. The size of the duplication varies from 100,000 to a few million DNA building blocks (base pairs). The MECP2 gene is always included in this duplication, and other genes may also be involved, depending on the size of the duplicated segment. It is unclear whether extra copies of these other genes affect the severity of the condition.
The MECP2 gene provides instructions for making a protein called MeCP2 that is critical for normal brain function. Researchers believe that this protein has several functions, including regulating other genes in the brain by controlling when they are able to participate in protein production. An extra copy of the MECP2 gene leads to the production of excess MeCP2 protein and an increase in protein function. The resulting changes in gene regulation and protein production in the brain lead to abnormal nerve cell (neuronal) function. These neuronal changes disrupt normal brain activity, causing the signs and symptoms of MECP2 duplication syndrome.

Inheritance
MECP2 duplication syndrome is inherited in an X-linked pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes in each cell. In males (who have only one X chromosome), a duplication of the only copy of the MECP2 gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a duplication of one of the two copies of the gene typically does not cause the disorder, but can be associated with neurodevelopmental problems such as depression, anxiety, and features of autism spectrum disorder that affect communication and social interaction.
in each cell. In males (who have only one X chromosome), a duplication of the only copy of the MECP2 gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a duplication of one of the two copies of the gene typically does not cause the disorder, but can be associated with neurodevelopmental problems such as depression, anxiety, and features of autism spectrum disorder that affect communication and social interaction.
Females with a MECP2 gene duplication tend to be unaffected or less severely affected than males because the X chromosome that contains the duplication may be turned off (inactive) in many of their cells due to a process called X-inactivation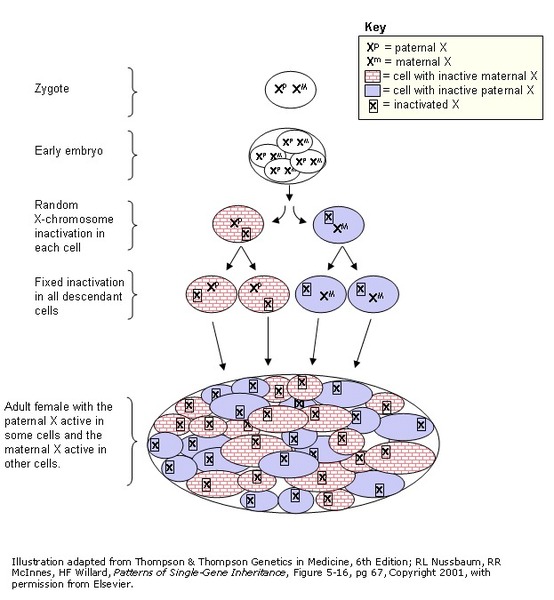 . Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body's cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
. Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body's cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
Females with a MECP2 gene duplication often have skewed X-inactivation, which results in the inactivation of the X chromosome containing the duplication in most cells of the body. Although this skewed X-inactivation ensures that the chromosome with the normal MECP2 gene is active most often, some of these females develop behavioral and psychiatric symptoms thought to be related to the additional genetic material. It is unclear why these features develop in a small number of females with skewed X-inactivation. Researchers speculate that in these females some cells in the brain may have a different pattern of X-inactivation than the cells in the rest of the body so that the X chromosome with the duplicated MECP2 gene is active, resulting in behavioral and psychiatric symptoms.
Other Names for This Condition
- Lubs X-linked mental retardation syndrome
- Trisomy Xq28
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- El Chehadeh S, Touraine R, Prieur F, Reardon W, Bienvenu T, Chantot-Bastaraud S, Doco-Fenzy M, Landais E, Philippe C, Marle N, Callier P, Mosca-Boidron AL, Mugneret F, Le Meur N, Goldenberg A, Guerrot AM, Chambon P, Satre V, Coutton C, Jouk PS, Devillard F, Dieterich K, Afenjar A, Burglen L, Moutard ML, Addor MC, Lebon S, Martinet D, Alessandri JL, Doray B, Miguet M, Devys D, Saugier-Veber P, Drunat S, Aral B, Kremer V, Rondeau S, Tabet AC, Thevenon J, Thauvin-Robinet C, Perreton N, Des Portes V, Faivre L. Xq28 duplication including MECP2 in six unreported affected females: what can we learn for diagnosis and genetic counselling? Clin Genet. 2017 Apr;91(4):576-588. doi: 10.1111/cge.12898. Epub 2017 Feb 16. Citation on PubMed
- Gonzales ML, LaSalle JM. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr Psychiatry Rep. 2010 Apr;12(2):127-34. doi: 10.1007/s11920-010-0097-7. Citation on PubMed or Free article on PubMed Central
- Lombardi LM, Baker SA, Zoghbi HY. MECP2 disorders: from the clinic to mice and back. J Clin Invest. 2015 Aug 3;125(8):2914-23. doi: 10.1172/JCI78167. Epub 2015 Aug 3. Citation on PubMed or Free article on PubMed Central
- Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J, Goizet C, Lacombe D, Pedespan JM, Echenne B, Tariverdian G, O'Rourke D, King MD, Green A, van Kogelenberg M, Van Esch H, Gecz J, Hamel BC, van Bokhoven H, de Brouwer AP. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009 Apr;17(4):444-53. doi: 10.1038/ejhg.2008.208. Epub 2008 Nov 5. Citation on PubMed or Free article on PubMed Central
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, Zoghbi HY. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009 Dec;66(6):771-82. doi: 10.1002/ana.21715. Citation on PubMed or Free article on PubMed Central
- Yi Z, Pan H, Li L, Wu H, Wang S, Ma Y, Qi Y. Chromosome Xq28 duplication encompassing MECP2: Clinical and molecular analysis of 16 new patients from 10 families in China. Eur J Med Genet. 2016 Jun;59(6-7):347-53. doi: 10.1016/j.ejmg.2016.05.004. Epub 2016 May 11. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.