

Description
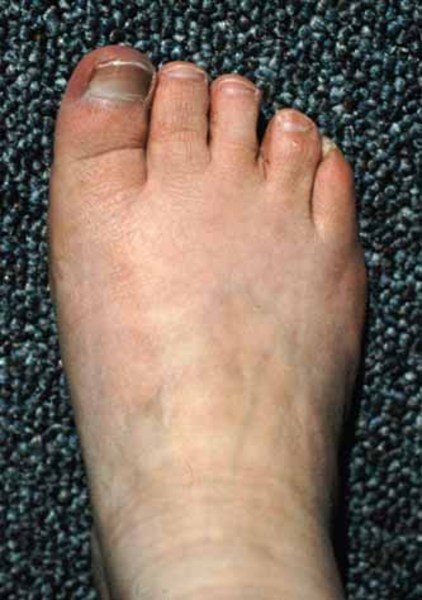
Mal de Meleda is a rare skin disorder that begins in early infancy. Affected individuals have a condition known as palmoplantar keratoderma, in which the skin of the palms of the hands and soles of the feet becomes thick, hard, and callused. In mal de Meleda, the thickened skin is also found on the back of the hands and feet and on the wrists and ankles. In addition, affected individuals may have rough, thick pads on the joints of the fingers and toes and on the elbows and knees. Some people with mal de Meleda have recurrent fungal infections in the thickened skin, which can lead to a strong odor. Other features of this disorder can include short fingers and toes (brachydactyly), nail abnormalities, red skin around the mouth, and excessive sweating (hyperhidrosis).

Frequency
Mal de Meleda is a rare disorder; its prevalence is unknown. The disorder was first identified on the Croatian island of Mljet (called Meleda in Italian) and has since been found in populations worldwide.
Causes
Mal de Meleda is caused by mutations in the SLURP1 gene. This gene provides instructions for making a protein that interacts with other proteins, called receptors, and is likely involved in signaling within cells. Studies show that the SLURP-1 protein can attach (bind) to nicotinic acetylcholine receptors (nAChRs) in the skin. Through interaction with these receptors, the SLURP-1 protein is thought to be involved in controlling the growth and division (proliferation), maturation (differentiation), and survival of skin cells.
Mutations in the SLURP1 gene lead to little or no SLURP-1 protein in the body. It is unclear how a lack of this protein leads to the skin problems that occur in mal de Meleda. Researchers speculate that without SLURP-1, the activity of genes controlled by nAChR signaling is altered, leading to overgrowth of skin cells or survival of cells that normally would have died. The excess of cells can result in skin thickening. It is unclear why skin on the hands and feet is particularly affected.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Acroerythrokeratoderma
- Keratosis palmoplantaris transgrediens of Siemens
- Meleda disease
- Transgrediens palmoplantar keratoderma of Siemens
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Arredondo J, Chernyavsky AI, Webber RJ, Grando SA. Biological effects of SLURP-1 on human keratinocytes. J Invest Dermatol. 2005 Dec;125(6):1236-41. doi: 10.1111/j.0022-202X.2005.23973.x. Citation on PubMed
- Chimienti F, Hogg RC, Plantard L, Lehmann C, Brakch N, Fischer J, Huber M, Bertrand D, Hohl D. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet. 2003 Nov 15;12(22):3017-24. doi: 10.1093/hmg/ddg320. Epub 2003 Sep 23. Citation on PubMed
- Eckl KM, Stevens HP, Lestringant GG, Westenberger-Treumann M, Traupe H, Hinz B, Frossard PM, Stadler R, Leigh IM, Nurnberg P, Reis A, Hennies HC. Mal de Meleda (MDM) caused by mutations in the gene for SLURP-1 in patients from Germany, Turkey, Palestine, and the United Arab Emirates. Hum Genet. 2003 Jan;112(1):50-6. doi: 10.1007/s00439-002-0838-8. Epub 2002 Oct 19. Citation on PubMed
- Favre B, Plantard L, Aeschbach L, Brakch N, Christen-Zaech S, de Viragh PA, Sergeant A, Huber M, Hohl D. SLURP1 is a late marker of epidermal differentiation and is absent in Mal de Meleda. J Invest Dermatol. 2007 Feb;127(2):301-8. doi: 10.1038/sj.jid.5700551. Epub 2006 Sep 28. Citation on PubMed
- Fischer J, Bouadjar B, Heilig R, Huber M, Lefevre C, Jobard F, Macari F, Bakija-Konsuo A, Ait-Belkacem F, Weissenbach J, Lathrop M, Hohl D, Prud'homme JF. Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum Mol Genet. 2001 Apr 1;10(8):875-80. doi: 10.1093/hmg/10.8.875. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.